1. Status of this document
This document provides information to the proteomics community about a proposed standard for sample metadata annotations in public repositories called Sample and Data Relationship Format (SDRF)-Proteomics. Distribution is unlimited.
Version v1.1.0 - 2026-01
2. Abstract
The Human Proteome Organisation (HUPO) Proteomics Standards Initiative (PSI) defines community standards for data representation in proteomics to facilitate data comparison, exchange, and verification. This document presents a specification for the Sample and Data Relationship Format (SDRF-Proteomics).
Further detailed information, including any updates to this document, implementations, and examples is available at SDRF GitHub Repository. The official PSI web page for the document is: HUPO-PSI SDRF.
3. Motivation
Public proteomics data is valuable, but sample metadata is often missing or stored inconsistently across repositories (e.g., CPTAC uses Excel files, ProteomicsDB captures minimal properties) [1]. This heterogeneity prevents reproducibility and cross-dataset integration.
SDRF-Proteomics addresses this by providing a standard tab-delimited format to capture (Figure 1):
-
Sample metadata and characteristics
-
Data file acquisition parameters
-
Sample-to-file relationships (experimental design)

Figure 1: SDRF-Proteomics captures sample information and its relationship to data files.
The format is fully compatible with MAGE-TAB SDRF, enabling integration with transcriptomics metadata standards.
4. Specification structure
SDRF-Proteomics uses a two-tier system: this core specification defines the format rules, and templates provide metadata checklists for specific experiment types (Figure 2). Templates are organized in the templates/ directory, each with documentation and example files.
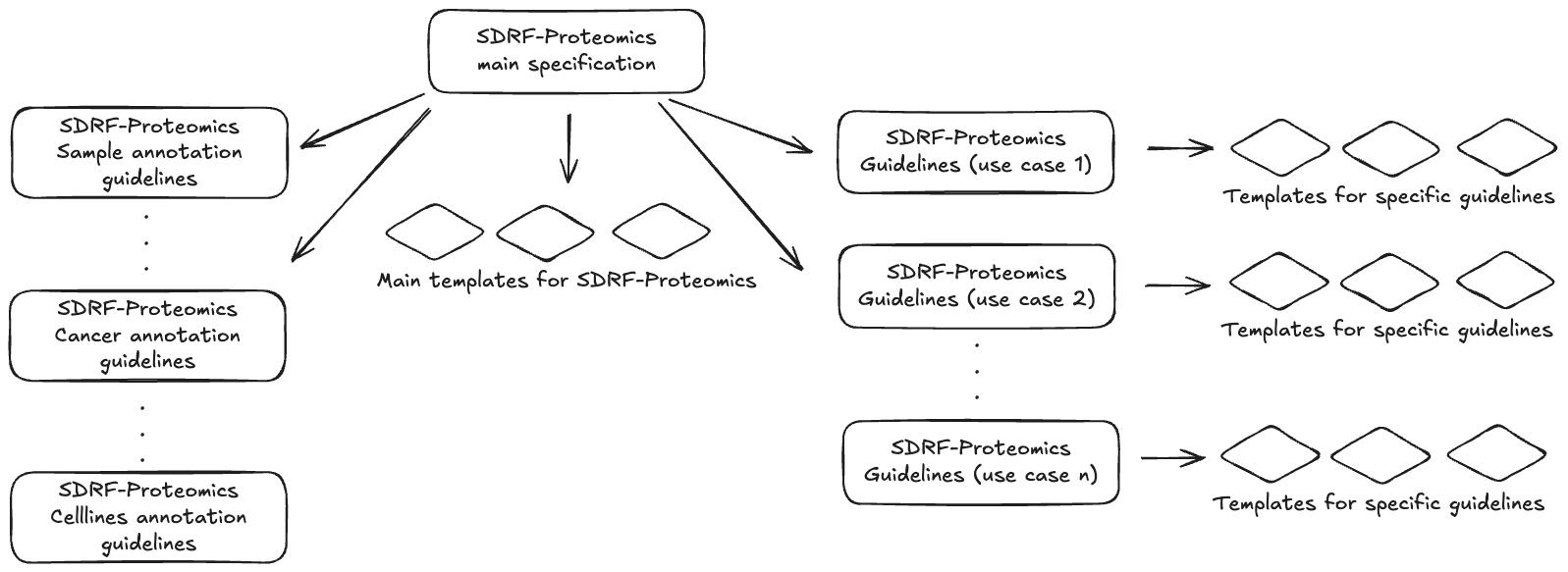
Figure 2: SDRF-Proteomics specification structure. The main specification defines the core rules and is extended by sample templates (human, vertebrates, etc.) and experiment-type templates (crosslinking, immunopeptidomics, etc.).
The official repository is GitHub, where you can find annotated example projects and the official validator sdrf-pipelines.
|
Important
|
Throughout this specification, the keywords "MUST", "REQUIRED", "SHOULD", "RECOMMENDED", and "OPTIONAL" are interpreted as described in RFC 2119. |
5. The SDRF-Proteomics Format
SDRF-Proteomics is a tab-delimited file where:
-
Each row = one sample linked to one data file
-
Each column = a property (sample characteristic, data file attribute, or factor value)
-
Each cell = the property value for that sample/file or a factor value.
Here’s a minimal example:
| source name | characteristics[organism] | characteristics[organism part] | characteristics[disease] | characteristics[biological replicate] | assay name | technology type | comment[proteomics data acquisition method] | comment[label] | comment[instrument] | comment[cleavage agent details] | comment[fraction identifier] | comment[technical replicate] | comment[data file] | factor value[disease] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
sample_1 |
homo sapiens |
liver |
normal |
1 |
run_1 |
proteomic profiling by mass spectrometry |
data-dependent acquisition |
label free sample |
Q Exactive HF |
NT=Trypsin;AC=MS:1001251 |
1 |
1 |
sample_1.raw |
normal |
sample_2 |
homo sapiens |
liver |
hepatocellular carcinoma |
1 |
run_2 |
proteomic profiling by mass spectrometry |
data-dependent acquisition |
label free sample |
Q Exactive HF |
NT=Trypsin;AC=MS:1001251 |
1 |
1 |
sample_2.raw |
hepatocellular carcinoma |
sample_3 |
homo sapiens |
not available |
not available |
1 |
run_3 |
proteomic profiling by mass spectrometry |
data-dependent acquisition |
label free sample |
Q Exactive HF |
NT=Trypsin;AC=MS:1001251 |
1 |
1 |
sample_3.raw |
not available |
The file is organized into three column sections:
-
Sample metadata (
characteristics[…]) - organism, disease, tissue, etc. -
Data file metadata (
comment[…]) - instrument, label, fraction, data file -
Factor values (
factor value[…]) - variables under study for statistical analysis
|
Note
|
|
5.1. Versioning
The SDRF-Proteomics specification uses Semantic Versioning (MAJOR.MINOR.PATCH). Version numbers are prefixed with "v" (e.g., v1.1.0). Changes are proposed via GitHub pull requests to the dev branch.
For the complete versioning strategy — including template versioning, ontology updates, the deprecation policy, transition timelines, and migration tooling — see Versioning and Deprecation Policy.
5.2. Format rules
-
Case sensitivity: Text values are case-insensitive, but column names are case-sensitive. Use lowercase for all column names (e.g.,
source name,characteristics[organism],comment[label]). Incorrect casing likeSource NameorCharacteristics[organism]will cause validation failures. -
Space sensitivity: The SDRF is sensitive to spaces in column names (
sourcename≠source name). Column names must include appropriate spaces (e.g.,source name, notsourcename) but must NOT have a space before the bracket (e.g.,characteristics[organism], notcharacteristics [organism]). -
Column order: The SDRF columns follows some structure; first the sample metadata columns in Chapter 7; then the data file metadata columns in Chapter 8; followed by the factor values columns in [study-variables].
-
Extension: The extension of the SDRF file SHOULD be sdrf.tsv (preferred) or .txt.
5.3. Reserved words
There are general scenarios where cell values cannot be provided with actual data. The following reserved words MUST be used in these cases. Reserved words MUST be all lowercase (e.g., not available, NOT Not Available or Not available):
-
not available: In some cases, the column is mandatory in the format, but for some samples the corresponding value is unknown or could not be determined. In those cases, users SHOULD use not available.
-
not applicable: In some cases, the column is mandatory, but for some samples the corresponding value or concept does not apply. In those cases, users SHOULD use not applicable.
-
anonymized: In some cases, the value exists but has been intentionally redacted for privacy protection (e.g., in clinical studies with de-identified patient data). In those cases, users SHOULD use anonymized.
-
pooled: In some cases, the sample is a pool of multiple samples (e.g., TMT reference channels), and the value cannot be represented as a single value. In those cases, users SHOULD use pooled.
| Term | Meaning | Example | Use Case |
|---|---|---|---|
not available |
Value exists but is unknown or could not be determined |
characteristics[age] = not available |
Patient age was not recorded in the study |
not applicable |
Value or concept does not apply to this sample |
characteristics[age] = not applicable |
Synthetic peptide library has no age |
anonymized |
Value exists but is redacted for privacy protection |
characteristics[age] = anonymized |
Clinical study with de-identified patient data |
pooled |
Value represents a mixture of multiple samples |
characteristics[biological replicate] = pooled |
TMT reference channel pooled from multiple replicates |
5.4. SDRF file-level metadata
Since version 1.1.0, SDRF-Proteomics supports file-level metadata using dedicated columns. These columns provide information about the SDRF file itself, such as the specification version, template(s) used, annotation tool, and validation status. This column-based approach maintains compatibility with spreadsheet applications (Excel, Google Sheets) and existing data processing tools.
The following metadata columns are supported:
| Column | Description | Example Value | Requirement | Ontology Term |
|---|---|---|---|---|
|
SDRF-Proteomics specification version used. Should follow semantic versioning format (vMAJOR.MINOR.PATCH) |
v1.1.0 |
RECOMMENDED |
PRIDE:0000839 |
|
Template name and version used for annotation. Two formats are supported: simple format ( |
human v1.1.0 or NT=human;VV=v1.1.0 |
OPTIONAL |
PRIDE:0000832 |
|
Software tool, script, or method used to generate or annotate the SDRF file. Two formats are supported: simple format ( |
lesSDRF v0.1.0 or NT=lesSDRF;VV=v0.1.0 |
OPTIONAL |
PRIDE:0000840 |
|
Cryptographic hash (e.g., SHA-256) generated after successful validation |
sha256:abc123… |
OPTIONAL |
PRIDE:0000834 |
|
Note
|
When combining multiple templates (e.g., human + ms-proteomics), use multiple comment[sdrf template] columns, one per template. The value in each row should be identical for all samples in the file.
|
Example of an SDRF file with metadata columns (simplified example showing only select columns; see Chapter 10 for complete required columns):
| source name | characteristics[organism] | characteristics[disease] | assay name | comment[data file] | comment[sdrf version] | comment[sdrf template] | comment[sdrf template] | comment[sdrf annotation tool] |
|---|---|---|---|---|---|---|---|---|
| sample_1 | homo sapiens | normal | run_1 | sample_1.raw | v1.1.0 | human v1.1.0 | ms-proteomics v1.1.0 | lesSDRF v0.1.0 |
| sample_2 | homo sapiens | breast cancer | run_2 | sample_2.raw | v1.1.0 | human v1.1.0 | ms-proteomics v1.1.0 | lesSDRF v0.1.0 |
5.5. Table Column headers
Depending on each section the column headers (property names) will be prefixed with the following prefixes:
-
characteristics: Sample metadata (e.g. characteristics[organism]) -
comment: Data file metadata (e.g. comment[data file]) -
factor value: Factor values properties (e.g. factor value[disease])
Each property name MUST be a valid ontology term or a valid controlled vocabulary term. Each section will have some specific order for column headers.
|
Note
|
A list of all controlled vocabularies and ontologies supported are in the Chapter 12 section. On each section we also provide a list of properties that are supported. |
5.6. Table Cell values
The value for each property, (e.g. characteristics, comment, factor value) corresponding to each sample or data file can be represented in multiple ways.
-
Free Text (Human readable): In the free text representation, the value is provided as text without Ontology support (e.g. colon or providing accession numbers). This is only RECOMMENDED when the text inserted in the table is the exact name of an ontology/CV term in EFO. If the term is not in EFO, other ontologies can be used.
| source name | characteristics[organism] |
|---|---|
sample 1 |
homo sapiens |
sample 2 |
homo sapiens |
-
Ontology url (Computer readable): Users can provide the corresponding URI (Uniform Resource Identifier) of the ontology/CV term as a value. This is recommended for enriched files where the user does not want to use intermediate tools to map from free text to ontology/CV terms.
-
Key=value representation (Human and Computer readable): The current representation aims to provide a mechanism to represent the complete information of the ontology/CV term including Accession, Name and other additional properties. In the key=value pair representation, the Value of the property is represented as an Object with multiple properties, where the key is one of the properties of the object and the value is the corresponding value for the particular key. The key order MUST be
NT(name) first, followed byAC(accession), then any additional keys. An example of key value pairs is post-translational modification (see Protein Modifications):NT=Glu->pyro-Glu;AC=Unimod:27;MT=fixed;PP=Anywhere;TA=E
|
Note
|
Beyond these three representations, SDRF columns may accept additional structured value types such as numbers with units (10 ppm), accession identifiers (SAMN12345678), ISO 8601 dates, semantic versions, and more. Each column’s YAML template definition declares exactly which value types and formats are accepted. For the complete reference of all value types, parsing rules, and their formal patterns, see Value Types Reference in the Templates Guide.
|
6. Validating SDRF Files
The official validator for SDRF-Proteomics files is sdrf-pipelines, a Python tool that checks your SDRF file for errors and compliance with the specification.
Installation:
pip install sdrf-pipelinesBasic Validation:
# Validate an SDRF file
parse_sdrf validate-sdrf --sdrf_file your_file.sdrf.tsv
# Validate with a specific template
parse_sdrf validate-sdrf --sdrf_file your_file.sdrf.tsv --template humanFor more information, visit: sdrf-pipelines on GitHub
7. SDRF-Proteomics: Samples metadata
The Sample metadata section provides information about the samples of origin and their characteristics. Each sample contains a source name (unique identifier) and a set of characteristics columns. The first column of the file should be the source name and the following columns should be the characteristics of the sample. For example, for any proteomics experiment (human, vertebrate, cell line), the following characteristics should be provided:
-
source name: Unique sample name (it can be present multiple times if the same sample is used several times in the same dataset)
-
characteristics[organism]: The organism of the Sample of origin. Values MUST come from NCBI Taxonomy.
-
characteristics[organism part]: The main normalized anatomical term for the sample (e.g., liver). Values SHOULD come from UBERON or BTO. Use the clearest ontology-backed anatomy term available for cross-study integration;
characteristics[tissue supergroup]can be used separately for broader organ/system grouping andcharacteristics[sampling site]for finer provenance.characteristics[sampling site]may be equal tocharacteristics[organism part]when no distinction exists. -
characteristics[disease]: The disease under study in the Sample. Values SHOULD come from MONDO, EFO, or DOID. For healthy/control samples, use
normal(PATO:0000461) - see Disease Annotation Guidelines. -
characteristics[cell type]: A cell type is a distinct morphological or functional form of cell (e.g., epithelial, glial). Values SHOULD come from Cell Ontology (CL), BTO, or Cell Line Ontology (CLO).
Example:
| source name | characteristics[organism] | characteristics[organism part] | characteristics[disease] | characteristics[cell type] |
|---|---|---|---|---|
sample_treat |
homo sapiens |
liver |
liver cancer |
not available |
sample_control |
homo sapiens |
liver |
liver cancer |
not available |
|
Note
|
|
7.1. BioSamples database integration
Use the OPTIONAL characteristics[biosample accession number] column to link samples to BioSamples [5], enabling cross-database integration with genomics and transcriptomics data. Formats: SAMN* (NCBI) or SAMEA* (EBI).
7.2. Encoding sample technical and biological replicates
SDRF-Proteomics uses two REQUIRED columns to track replicates [4]:
-
characteristics[biological replicate]: Independent biological samples. Numbering restarts per experimental condition (factor value group).
-
comment[technical replicate]: Repeated measurements of the same sample (e.g., multiple injections)
When no replicates are performed, set both columns to 1. For pooled samples, use pooled for biological replicate.
| source name | characteristics[biological replicate] | comment[fraction identifier] | comment[technical replicate] | comment[data file] |
|---|---|---|---|---|
patient_001 |
1 |
1 |
1 |
P001_F1_TR1.raw |
patient_001 |
1 |
1 |
2 |
P001_F1_TR2.raw |
patient_002 |
2 |
1 |
1 |
P002_F1_TR1.raw |
patient_002 |
2 |
1 |
2 |
P002_F1_TR2.raw |
7.3. Pooled samples
When multiple samples are pooled into one (e.g., TMT/iTRAQ reference channels for normalization), use the characteristics[pooled sample] column to indicate pooling status. Allowed values:
-
not pooled: Regular individual samples
-
pooled: Sample is pooled but individual sources are unknown
-
SN=sample1;SN=sample2;…: Lists source names of pooled samples when known
Example:
| source name | characteristics[pooled sample] | characteristics[organism] | characteristics[age] | comment[label] | comment[data file] |
|---|---|---|---|---|---|
sample_1 |
not pooled |
homo sapiens |
45Y |
TMT126 |
file01.raw |
sample_2 |
not pooled |
homo sapiens |
52Y |
TMT127N |
file01.raw |
pooled_ref |
SN=sample_1;SN=sample_2 |
homo sapiens |
pooled |
TMT131C |
file01.raw |
|
Tip
|
For pooled samples, use pooled for individual-specific fields (biological replicate, age, sex) to indicate a mixture rather than a single sample.
|
7.4. Sample Metadata Guidelines
For detailed guidance on annotating sample metadata, refer to the following conventions documents:
-
Sample Metadata Guidelines - Detailed guidelines for age, sex, disease, organism part, cell type, developmental stage, spiked-in samples, and other sample characteristics
-
Human Sample Metadata Guidelines - Human-specific metadata including disease staging, treatment history, demographics, and lifestyle factors
8. SDRF-Proteomics: data files metadata
The connection between samples and data files is done using properties annotated with the comment prefix. All properties referring to a data file (e.g., MS run file) are annotated with the category comment. This differentiates data file properties from sample properties (characteristics).
8.1. CV Term Format for Data File Metadata
For data file metadata (comment columns) that reference ontology terms, use the structured format: NT={term name};AC={accession}
Examples: NT=HCD;AC=PRIDE:0000590, NT=Orbitrap;AC=MS:1000484
This format enables automated validation and software extraction from raw files. Sample metadata (characteristics) can use simple term names since they are typically human-annotated.
The following properties MUST be provided for each data file in mass spectrometry-based proteomics experiments. For affinity-based proteomics (Olink, SomaScan), see the Affinity-Proteomics template for different required columns.
| Column | Requirement | Description | Ontology |
|---|---|---|---|
|
REQUIRED |
Unique identifier for an MS run/data file |
Free text |
|
REQUIRED |
Technology used to capture the data |
Fixed values |
|
REQUIRED |
DDA, DIA, PRM, SRM |
PRIDE:0000659 |
|
REQUIRED |
Label applied to sample (or "label free sample") |
PRIDE - Labels |
|
REQUIRED |
Mass spectrometer model |
PSI-MS - Instruments |
|
REQUIRED |
Enzyme information (use "not applicable" for top-down/undigested samples) |
PSI-MS - Cleavage agents |
|
REQUIRED |
Fraction number (1 if not fractionated) |
Integer |
|
REQUIRED |
Technical replicate number (1 if none) |
Integer |
|
REQUIRED |
Name of the raw file (primary/canonical file when the vendor format ships multiple files - see Section 8.2) |
Free text |
Example:
| source name | assay name | technology type | comment[proteomics data acquisition method] | comment[label] | comment[instrument] | comment[data file] |
|---|---|---|---|---|---|---|
sample_1 |
sample1_run1 |
proteomic profiling by mass spectrometry |
data-dependent acquisition |
label free sample |
Q Exactive HF |
sample1.raw |
8.2. Vendor formats that ship multiple files (e.g. AB Sciex .wiff + .wiff.scan)
Some vendor formats persist a single MS run as two or more files on disk that are only useful together. The most common case is AB Sciex, where every acquisition produces a .wiff file paired with a .wiff.scan (and sometimes .wiff2) sidecar; tools such as ProteoWizard/Skyline cannot open one without the other. Bruker .d directories, in contrast, are a single logical entity (a folder) and do not require this pattern - reference the .d folder name directly in comment[data file].
To represent these multi-file formats while keeping one row per MS run, SDRF-Proteomics treats one file as the primary/canonical file and the remaining files as auxiliary files described by sidecar columns:
| Column | Cardinality | Description |
|---|---|---|
|
1 |
Name of the primary/canonical file for the MS run (for AB Sciex, the |
|
1 |
URI to retrieve the primary file. The trailing path segment MUST equal |
|
* |
Name of an auxiliary file that must be downloaded alongside the primary file (for AB Sciex, the |
|
* |
URI to retrieve the auxiliary file. The trailing path segment MUST equal the corresponding |
|
Note
|
When multiple comment[associated data file] / comment[associated file uri] columns are present, consumers MUST pair them positionally (first associated file with first associated URI, second with second, etc.).
|
Example - AB Sciex (.wiff + .wiff.scan):
| source name | assay name | comment[instrument] | comment[data file] | comment[file uri] | comment[associated data file] | comment[associated file uri] |
|---|---|---|---|---|---|---|
sample_1 |
sample1_run1 |
TripleTOF 6600 |
OA_3.wiff |
https://ftp.pride.ebi.ac.uk/pride/data/archive/2026/03/PXD073289/OA_3.wiff |
OA_3.wiff.scan |
https://ftp.pride.ebi.ac.uk/pride/data/archive/2026/03/PXD073289/OA_3.wiff.scan |
|
Tip
|
Once repositories standardise on bundling related files into a single .zip or .tar.gz (as already done for some Bruker timsTOF deposits), comment[data file] and comment[file uri] can reference the archive directly and comment[associated data file] is no longer needed. See the PRIDE submission formats guidelines for the direction of travel.
|
If a repository already distributes a single canonical archive for one acquisition today (for example a .raw.zip bundle in some metabolomics deposits), that archive can already be recorded directly in comment[data file] and comment[file uri].
8.3. Sample Preparation and Fragmentation (MS-based only)
|
Note
|
This section applies to mass spectrometry-based proteomics experiments only. For affinity-based proteomics, these properties do not apply. |
For detailed documentation of sample preparation and MS/MS fragmentation properties, see the MS-Proteomics Template:
-
Sample preparation: depletion, reduction reagent, alkylation reagent
-
Fractionation: fractionation method (used with
comment[fraction identifier]) -
Fragmentation: collision energy, dissociation method
|
Note
|
For HCD (Higher-energy C-trap Dissociation), the canonical accession is MS:1000422 - beam-type collision-induced dissociation. Use NT=beam-type collision-induced dissociation;AC=MS:1000422 or the short label HCD. Do not use PRIDE:0000590 or MS:1002481.
|
8.4. Proteomics data acquisition method
Proteomics data acquisition method can happen in multiple ways: Data Dependent Acquisition (DDA), Data Independent Acquisition (DIA), and targeted approaches. The SDRF-Proteomics file format REQUIRES capturing the method used for the data acquisition in the comment[proteomics data acquisition method] column. The values MUST be children of the PRIDE ontology term proteomics data acquisition method (PRIDE:0000659). The following values are commonly used:
|
Important
|
The comment[proteomics data acquisition method] column is REQUIRED for all mass spectrometry-based SDRF files. This field must be explicitly specified and cannot be omitted or assumed. |
You can find an example of a DIA experiment in the following link: DIA example
|
Tip
|
For DIA experiments, additional properties like MS1 scan range can be captured. See DIA Scan Window Limits in the DIA-Acquisition Template. |
8.5. MS-Proteomics Template
For detailed guidance on data file metadata, refer to the conventions document:
-
MS-Proteomics Template - Detailed guidelines for labels, instruments, modifications, cleavage agents, mass tolerances, RAW file URIs, and other data file properties
9. Additional SDRF Rules
9.1. Column Cardinality
Some columns can appear multiple times for the same sample. The cardinality rules are:
-
Single (1): Column appears exactly once per sample (e.g.,
characteristics[biological replicate]) -
Multiple (*): Column can appear multiple times (e.g.,
comment[modification parameters]can specify multiple post-translational modifications)
Example of multiple comment[modification parameters] columns:
| source name | characteristics[…] | comment[modification parameters] | comment[modification parameters] | … |
|---|---|---|---|---|
sample-1 |
… |
NT=Carbamidomethyl;AC=UNIMOD:4;TA=C;MT=fixed;PP=Anywhere |
NT=Oxidation;AC=UNIMOD:35;TA=M;MT=variable;PP=Anywhere |
… |
9.2. Row Uniqueness Requirements
Uniqueness constraints ensure data integrity:
-
MUST be unique (error):
source name+assay name+comment[label] -
SHOULD be unique (warning):
source name+assay name -
Assay name: Each data file MUST have a unique
assay name
|
Note
|
For multiplexed experiments (TMT, iTRAQ), multiple rows share the same assay name since samples are in one MS run. The comment[label] distinguishes samples within the run.
|
10. Templates
A template is a predefined set of metadata columns that ensures consistent annotation for specific experiment types. Templates define REQUIRED, RECOMMENDED, and OPTIONAL columns to make datasets FAIR-compliant.
10.1. Template Architecture
Templates follow a layered hierarchy:
| Layer | Templates | Description |
|---|---|---|
TECHNOLOGY (required) |
Minimum valid SDRF - choose one |
|
SAMPLE (recommended) |
human, vertebrates, invertebrates, plants, clinical-metadata, oncology-metadata |
Organism-specific and clinical metadata |
EXPERIMENT (optional) |
cell-lines, crosslinking, dia-acquisition, single-cell, immunopeptidomics, metaproteomics, lc-ms-metabolomics, gc-ms-metabolomics |
Methodology-specific columns |
Child templates inherit all columns from parents and may add new columns or strengthen requirements (e.g., optional → required).
10.2. Template Combination Rules
Some layers enforce mutually exclusive choices, while others allow combining multiple templates:
| Layer | Templates | Rule |
|---|---|---|
TECHNOLOGY |
|
Mutually exclusive — choose one (REQUIRED) |
SAMPLE |
|
Mutually exclusive — choose one based on organism (RECOMMENDED) |
EXPERIMENT (MS) |
|
Can be combined (e.g., |
EXPERIMENT (metabolomics) |
|
Mutually exclusive — choose one when using |
Templates from different layers can be freely combined. Common valid combinations:
-
ms-proteomics+human(human DDA proteomics) -
ms-proteomics+human+dia-acquisition(human DIA proteomics) -
ms-proteomics+human+immunopeptidomics(human immunopeptidomics) -
ms-proteomics+vertebrates+cell-lines(mouse cell line proteomics) -
ms-proteomics+human+crosslinking(human crosslinking MS) -
affinity-proteomics+human(human affinity proteomics, including Olink or SomaScan) -
ms-metabolomics+human+lc-ms-metabolomics(human LC-MS metabolomics) -
ms-metabolomics+plants+gc-ms-metabolomics(plant GC-MS metabolomics) -
ms-proteomics+metaproteomics(environmental metaproteomics) -
ms-proteomics+human-gut(human gut microbiome metaproteomics) -
ms-proteomics+human+single-cell(human single-cell proteomics)
10.3. Specifying Templates in SDRF Files
Declare templates using comment[sdrf template] columns. Only list leaf templates (parents are implied). When using multiple templates, add multiple columns with the same name. Two formats are supported:
-
Simple format (preferred):
template_name vX.Y.Z -
Key=value format:
NT=template_name;VV=vX.Y.Z
source name ... comment[sdrf template] comment[sdrf template]
sample_1 ... human v1.1.0 crosslinking v1.0.0Common examples:
| Experiment Type | Template Columns |
|---|---|
Human MS proteomics |
|
Mouse MS proteomics |
|
Human crosslinking |
Two columns: |
Human affinity proteomics |
Two columns: |
Human LC-MS metabolomics |
Two columns: |
10.4. Available Templates
Sample templates (organism-specific):
| Template | Use For | Key Columns |
|---|---|---|
Human clinical samples |
disease, age, sex, ancestry |
|
Mouse, rat, zebrafish |
disease, developmental stage, strain |
|
Drosophila, C. elegans |
disease, developmental stage, genotype |
|
Arabidopsis, crops |
disease, developmental stage, growth conditions |
Experiment-type templates:
-
Affinity Proteomics - Olink and SomaScan-style platform metadata
-
DIA Acquisition - scan windows, isolation width
-
Cell Lines - Cellosaurus integration
-
Single-Cell - cell isolation, carrier proteome
-
Immunopeptidomics - MHC protein complex, MHC typing
-
Crosslinking MS - crosslinker reagents
-
LC-MS Metabolomics - chromatography, ion source, polarity, scan range
-
GC-MS Metabolomics - derivatization, GC column, carrier gas, oven program
-
Metaproteomics - environmental sample type
Download templates from the templates folder.
10.5. Extending Templates
You can add custom columns beyond template requirements for study-specific metadata. Rules:
-
Use
characteristics[…]for sample metadata,comment[…]for technical metadata -
Column names MUST be valid ontology terms (search OLS)
-
Use controlled vocabularies for values when available
See Additional Sample-Related Columns and SDRF Terms Reference for commonly used columns.
10.6. Contributing New Templates
To propose a new template, open an issue on GitHub and submit a pull request.
11. Factor Values (Study Variables)
Factor values identify the experimental variables being studied - the conditions you want to compare in your analysis. They highlight which sample characteristics are the focus of your experiment.
11.1. Column Format
factor value[{variable name}]
11.2. When to Use Factor Values
Use factor values to indicate:
-
The primary variable(s) under investigation
-
Conditions being compared (e.g., disease vs. normal, treated vs. untreated)
-
Variables that define experimental groups
|
Note
|
Use normal (not "control") in the disease field for healthy samples. "Control" is an experimental design concept, not a disease state. See Disease Annotation Guidelines for details.
|
11.3. Rules
-
Factor value columns SHOULD appear after all characteristics and comment columns
-
Multiple factor values can be used when studying multiple variables
-
The value in a factor value column typically mirrors a characteristics column value
11.4. Example
In an experiment comparing tumor vs. normal tissue across different cancer stages:
| source name | … | characteristics[disease] | characteristics[disease staging] | … | factor value[disease] | factor value[disease staging] |
|---|---|---|---|---|---|---|
tumor_sample_1 |
… |
breast carcinoma |
stage II |
… |
breast carcinoma |
stage II |
normal_sample_1 |
… |
normal |
not applicable |
… |
normal |
not applicable |
tumor_sample_2 |
… |
breast carcinoma |
stage III |
… |
breast carcinoma |
stage III |
In this example, both disease and disease staging are factor values because the experiment aims to compare expression differences between disease states and across cancer stages.
12. Ontologies and Controlled Vocabularies
SDRF-Proteomics uses ontologies and controlled vocabularies (CVs) to standardize metadata values. The following ontologies are supported:
| Category | Ontology/CV | Description | Notes |
|---|---|---|---|
General Purpose |
|||
General |
General experimental metadata |
||
General |
Phenotype and Trait Ontology |
||
General |
Biomedical terminology |
||
General |
Proteomics-specific terms |
||
Organism and Taxonomy |
|||
Taxonomy |
Organism classification |
||
Anatomy and Cell Types |
|||
Anatomy |
Cross-species anatomy ontology |
||
Cell Type |
Cell type classification |
||
Anatomy |
Tissues and cell lines |
||
Anatomy |
Plant anatomy and development |
For plant samples |
|
Anatomy |
Drosophila anatomy |
For Drosophila samples |
|
Anatomy |
C. elegans anatomy |
For C. elegans samples |
|
Anatomy |
Zebrafish anatomy and development |
For zebrafish samples |
|
Disease (see Disease Annotation Guidelines) |
|||
Disease |
Unified disease ontology |
RECOMMENDED |
|
Disease |
Disease terms from EFO |
||
Healthy samples |
Use |
||
Cell Lines |
|||
Cell Lines |
Cell line knowledge resource |
RECOMMENDED |
|
Cell Lines |
Cell line ontology |
||
Mass Spectrometry and Proteomics |
|||
MS/Proteomics |
Instruments, methods, parameters |
||
Modifications |
Protein modifications database |
||
Modifications |
Protein modifications ontology |
||
Other |
|||
Chemistry |
Chemical Entities of Biological Interest |
||
Environment |
Environmental sample classification |
For metaproteomics |
|
Ancestry |
Human ancestry categories |
For human samples |
|
13. Examples of Annotated Datasets
The following table provides links to example SDRF files for different experiment types. Click "View in Explorer" to open the SDRF file in the interactive viewer.
| Experiment Type | Dataset | Description | View | Source |
|---|---|---|---|---|
Label-free |
PXD008934 |
Human proteome label-free quantification |
||
TMT |
PXD017710 |
TMT-labeled quantitative proteomics |
||
SILAC |
PXD000612 |
SILAC-based quantification |
||
DIA |
PXD018830 |
data-independent acquisition |
||
Phosphoproteomics |
PXD000759 |
PTM enrichment study |
||
Cell lines |
PXD001819 |
Cell line proteomics |
|
Tip
|
Use the SDRF Explorer to browse all {total_datasets}+ annotated datasets with filtering, statistics, and interactive viewing. |
A comprehensive collection of annotated projects is available at: Annotated Projects Repository
14. Template Definitions
This section provides the column definitions for each SDRF-Proteomics template. Each template shows only its own columns (not inherited ones). See the "Extends" field to identify which parent template’s columns are also included.
14.1. base
Version: 1.1.0 | Layer: internal | Extends: none | Usable alone: No
Base SDRF template with infrastructure columns (identifiers, data files, versioning) inherited by all proteomics templates. This is a construction artifact and cannot be used directly.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Unique identifier for the biological sample |
||
|
required |
Unique identifier for the data acquisition run |
||
|
required |
Type of technology used |
single value only; values: proteomic profiling by mass spectrometry, protein expression profiling by antibody array, protein expression profiling by aptamer array |
|
|
required |
Identifier for the technical replicate (integer starting from 1) |
||
|
required |
Name of the raw data file |
||
|
recommended |
Version of the SDRF-Proteomics specification used to annotate this file |
semver |
v1.1.0, v2.0.0-dev |
|
optional |
Template name and version used for annotation. Two formats are supported - key=value format (NT=template_name;VV=vX.Y.Z) or simple format (template_name vX.Y.Z). Multiple templates can be specified using multiple columns. |
pattern: Template can be specified as 'NT=name;VV=vX.Y.Z' or 'name vX.Y.Z' |
NT=human;VV=v1.1.0, human v1.1.0, NT=ms-proteomics;VV=v1.1.0, ms-proteomics v1.1.0 |
|
optional |
Software tool or method used to generate or annotate the SDRF file. Two formats are supported - key=value format (NT=tool_name;VV=vX.Y.Z) or simple format (tool_name vX.Y.Z). |
pattern: Annotation tool can be specified as 'NT=name;VV=vX.Y.Z' or 'name vX.Y.Z' or 'manual curation' |
NT=lesSDRF;VV=v0.1.0, lesSDRF v0.1.0, NT=sdrf-pipelines;VV=v1.0.0, sdrf-pipelines v1.0.0, … |
|
optional |
Hash value for SDRF validation integrity checking |
pattern: Validation hash string |
14.2. sample-metadata
Version: 1.0.0 | Layer: internal | Extends: base | Usable alone: No
SDRF template with shared sample metadata columns (organism, tissue, disease). This is an internal construction layer inherited by technology and organism templates - not used directly.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Species of the sample using NCBI Taxonomy |
ontology: ncbitaxon |
homo sapiens, mus musculus, rattus norvegicus, saccharomyces cerevisiae |
|
required |
Main normalized anatomical term for the sample |
ontology: uberon, bto |
liver, brain, heart, blood |
|
optional |
Broader anatomical grouping or system-level bucket for the sample, used alongside the organism part for higher-level grouping |
ontology: uberon, bto |
digestive system, nervous system, cardiovascular system, gastrointestinal tract |
|
recommended |
Cell type of the sample |
ontology: cl, bto, clo |
hepatocyte, neuron, fibroblast, T cell |
|
required |
Identifier for the biological replicate (integer starting from 1, or 'pooled' for pooled samples) |
pattern: Biological replicate should be an integer or 'pooled' for pooled reference samples |
1, 2, pooled |
|
optional |
Whether the sample is a pooled sample combining material from multiple biological sources. Use 'not pooled' for individual samples, 'pooled' when sources are unknown, or 'SN=sample1;SN=sample2' to list source names. |
values: not pooled, pooled; pattern: Use 'not pooled', 'pooled', or list sample IDs with SN= prefix |
SN=sample1;SN=sample2 |
|
optional |
Classification of the sample role in the experiment. Distinguishes study samples under investigation from controls, references, and other roles in multiplexed or plate-based experiments. |
ontology: pride |
study sample, single cell, reference, bridge, carrier, negative control, positive control, calibrator, plate control, quality control sample, … |
|
recommended |
Disease state of the sample |
ontology: mondo, efo, doid, ncit, pato |
normal, breast cancer, infection, metabolic disease |
|
optional |
Type of biological material being analyzed |
values: tissue, cell, cell line, organism part, … |
|
|
optional |
Mass of tissue used for extraction |
number with unit (mg, g, ug) |
50 mg, 1 g, 500 ug |
|
optional |
BioSample accession number for the sample (e.g., SAMN or SAMEA identifiers) |
accession: biosample |
SAMN12345678, SAMEA12345678, SAMD1234567 |
|
optional |
Time at which the sample was collected (for longitudinal or time-course studies) |
number with unit (hour, day, minute, week, month, year) |
0 hour, 24 hour, 7 day, 3 month |
|
optional |
Treatment or perturbation applied to the sample (drug, stimulus, environmental stress) |
ontology: ncit, efo |
untreated, LPS stimulation, doxorubicin treatment, drought stress, … |
|
optional |
Whether the sample is a synthetic peptide library or biological material |
values: synthetic, not synthetic |
|
|
optional |
Spiked-in compound details using key-value format (CT=compound type, QY=quantity, PS=peptide sequence, AC=UniProt accession, CN=compound name, CV=vendor) |
pattern: Key-value format for spiked compound details (CT=type, SP=species, QY=quantity, PS=sequence, AC=accession, CN=name, CV=vendor) |
CT=peptide;PS=PEPTIDESEQ;QY=10 fmol, CT=protein;AC=A9WZ33;QY=20 nmol, CT=protein;SP=Homo sapiens;QY=1 pmol;AC=P37840, CT=mixture;CN=iRT mixture;CV=Biognosys;QY=1 pmol |
|
optional |
Enrichment strategy applied to the sample (e.g., phosphopeptide enrichment, crosslinked peptide enrichment, glycopeptide enrichment) |
ontology: pride, efo |
enrichment of cross-linked peptides, enrichment of phosphorylated protein, enrichment of glycopeptides, enrichment of ubiquitinated proteins |
14.3. ms-proteomics
Version: 1.1.0 | Layer: technology | Extends: sample-metadata | Usable alone: Yes
Base SDRF template for mass spectrometry-based proteomics. This is the minimum valid template for any MS experiment.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Mass spectrometry acquisition method |
ontology: pride |
data-dependent acquisition, data-independent acquisition, parallel reaction monitoring, selected reaction monitoring |
|
required |
Mass spectrometer instrument used |
ontology: ms, pride |
LTQ Orbitrap, Q Exactive, Orbitrap Fusion Lumos, timsTOF Pro |
|
required |
Enzyme or chemical used for protein digestion |
ontology: ms |
NT=Trypsin;AC=MS:1001251, NT=Lys-C;AC=MS:1001309, NT=Chymotrypsin;AC=MS:1001306 |
|
required |
Labeling strategy used for quantification |
ontology: pride |
label free sample, SILAC light, SILAC heavy, TMT126, … |
|
required |
Fraction number for fractionated samples (integer, use 1 for non-fractionated). In MS proteomics, this identifies the chromatographic or electrophoretic fraction (e.g., SCX, hpHRP, SEC fractions). Each fraction maps to one data file. |
||
|
recommended |
Fragmentation method used in MS/MS |
ontology: ms, pride |
HCD, CID, ETD, EThcD |
|
optional |
Peptide fractionation method used before MS analysis |
ontology: pride |
High-pH reversed-phase chromatography (hpHRP), Strong cation-exchange chromatography (SCX), Strong anion-exchange chromatography (SAX), Size-exclusion chromatography (SEC) |
|
optional |
Collision energy used for fragmentation |
pattern: Collision energy format: {value} {unit} where unit is NCE or eV. For multiple values, use semicolon-separated entries. |
30 NCE, 30% NCE, 27 eV, 25 NCE;27 NCE;30 NCE |
|
recommended |
Precursor mass tolerance for database search |
number with unit (ppm, Da, mmu) |
10 ppm, 20 ppm, 0.5 Da, 20 mmu |
|
recommended |
Fragment mass tolerance for database search |
number with unit (ppm, Da, mmu) |
0.02 Da, 20 ppm, 50 mmu |
|
optional |
Chemical reagent used for disulfide bond reduction |
ontology: pride, ms |
dithiothreitol, tris(2-carboxyethyl)phosphine |
|
optional |
Chemical reagent used for cysteine alkylation |
ontology: pride, ms |
iodoacetamide, chloroacetamide |
|
optional |
Whether abundant protein depletion was performed |
values: no depletion, depletion |
|
|
recommended |
Post-translational modifications searched |
ontology: unimod, mod |
NT=Oxidation;MT=Variable;TA=M;AC=Unimod:35, NT=Carbamidomethyl;TA=C;MT=fixed;AC=UNIMOD:4 |
|
optional |
Mass analyzer used for MS2 acquisition |
ontology: ms |
orbitrap, ion trap, TOF |
|
optional |
Batch identifier for sample preparation (plate, chip, processing batch). Useful for batch effect correction in multi-batch experiments. |
pattern: Sample preparation batch identifier |
plate1, batch_20220601, prep_A |
|
optional |
Liquid chromatography batch identifier for batch effect tracking (e.g., column changes, LC system swaps) |
pattern: LC batch identifier |
LC1, column_A |
|
optional |
Date of MS data acquisition (ISO 8601 format recommended). Useful for tracking instrument drift and batch effects. |
pattern: Acquisition date/time |
2022-06-01, 2022-06-01T18:28:37 |
|
optional |
MS method-defined minimum precursor (MS1) m/z setting used to acquire the data |
m/z value |
100m/z, 200m/z, 350.5m/z |
|
optional |
MS method-defined maximum precursor (MS1) m/z setting used to acquire the data |
m/z value |
1200m/z, 1600m/z, 2000m/z |
|
optional |
MS method-defined minimum precursor charge state setting used to acquire the data |
pattern: Integer charge state |
1, 2 |
|
optional |
MS method-defined maximum precursor charge state setting used to acquire the data |
pattern: Integer charge state |
6, 7, 8 |
|
optional |
LC method-defined minimum retention time setting used to acquire the data (in minutes) |
pattern: Numeric retention time in minutes |
0, 5, 10.5 |
|
optional |
LC method-defined maximum retention time setting used to acquire the data (in minutes) |
pattern: Numeric retention time in minutes |
60, 90, 120 |
|
optional |
MS method-defined minimum ion mobility setting used to acquire the data (1/K0 or Vs/cm2) |
pattern: Numeric ion mobility value |
0.6, 0.7 |
|
optional |
MS method-defined maximum ion mobility setting used to acquire the data (1/K0 or Vs/cm2) |
pattern: Numeric ion mobility value |
1.3, 1.4, 1.6 |
|
optional |
MS method-defined minimum product ion (MS2) m/z setting used to acquire the data |
m/z value |
100m/z, 200m/z |
|
optional |
MS method-defined maximum product ion (MS2) m/z setting used to acquire the data |
m/z value |
1800m/z, 2000m/z |
|
optional |
MS method-defined minimum product ion (MS3) m/z setting used to acquire the data |
m/z value |
100m/z, 200m/z |
|
optional |
MS method-defined maximum product ion (MS3) m/z setting used to acquire the data |
m/z value |
1500m/z, 2000m/z |
|
optional |
m/z scan range for MS1 spectra as an interval. Alternative to separate ms min mz / ms max mz columns |
m/z range interval |
400m/z-1200m/z, 350m/z-1600m/z |
|
optional |
m/z scan range for MS2 spectra as an interval. Alternative to separate ms2 min mz / ms2 max mz columns |
m/z range interval |
100m/z-2000m/z, 200m/z-1800m/z |
|
optional |
m/z scan range for MS3 spectra as an interval. Alternative to separate ms3 min mz / ms3 max mz columns |
m/z range interval |
100m/z-1500m/z, 200m/z-2000m/z |
|
optional |
Conditions used for peptide/protein elution |
pattern: Free-text elution conditions |
0.1% TFA in water, 80% acetonitrile, gradient 5-35% ACN in 60 min |
14.4. affinity-proteomics
Version: 1.0.0 | Layer: technology | Extends: sample-metadata | Usable alone: Yes
SDRF template for affinity-based proteomics experiments (Olink, SomaScan). This is the base template for all affinity proteomics experiments.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Role of the sample in the assay. Distinguishes study samples from controls and reference materials for assay interpretation and quality control |
ontology: pride |
study sample, negative control, positive control, calibrator, plate control, quality control sample, buffer control, bridge, reference, standard |
|
required |
Affinity proteomics platform used (e.g. Olink Explore HT, SomaScan Assay 7K) |
single value only; ontology: pride |
Olink Explore HT, Olink Target 96, SomaScan Assay 11K |
|
optional |
Instrument used for data acquisition (e.g. sequencer, qPCR machine, microarray reader) |
ontology: ms, pride |
Illumina NovaSeq X, Illumina NextSeq 2000, Agilent SureScan Microarray Scanner |
|
recommended |
Commercial panel/menu identifier within the platform. RECOMMENDED, but MUST be provided when not uniquely inferable from comment[platform]. |
pattern: Panel/menu identifier |
Target 96 Inflammation, Explore 1536, SomaScan 7K, SomaScan 11K |
|
optional |
Unit of quantification for the assay (platform-specific) |
values: NPX, RFU |
|
|
optional |
Plate identifier for batch effect analysis |
pattern: Plate identifier |
1, 2 |
|
optional |
Normalization method applied to quantification values |
pattern: Normalization method |
plate control normalized, bridge normalized, median normalization, not normalized |
|
optional |
Sample dilution factor used (commonly used for SomaScan workflows) |
pattern: Dilution factor; SomaScan commonly uses 0.005%, 0.5%, 20%, 40% |
0.005%, 0.5%, 20%, 40%, … |
|
optional |
Reagent lot number for traceability |
pattern: Lot number |
SS-2023-001, lot_12345 |
14.5. human
Version: 1.1.0 | Layer: sample | Extends: sample-metadata | Usable alone: No
Human SDRF template with human-specific sample metadata fields. Must be combined with a technology template (ms-proteomics, affinity-proteomics, or ms-metabolomics).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
(override: requirement set to required) |
||
|
recommended |
Ancestry or ethnic background of the donor |
ontology: hancestro |
European, African, Asian, Hispanic or Latin American |
|
required |
Age of the donor at sample collection |
pattern: Age format: 45Y, 6M, 30Y6M (Y>M>W>D order), ranges like 40Y-50Y, or comparison operators like >18Y, >=21Y, <65Y. Use "not available" if unknown, "anonymized" if redacted, or "pooled" for pooled samples. |
45Y, 6M, 30Y6M, 30Y6M2W, … |
|
required |
Biological sex of the donor |
values: male, female, intersex |
|
|
optional |
Developmental stage of the donor |
ontology: efo |
adult, embryonic stage, fetal stage, infant stage |
|
recommended |
Unique identifier for the donor individual |
identifier |
patient_001, donor-A1, subject_12, anonymized, … |
14.6. vertebrates
Version: 1.1.0 | Layer: sample | Extends: sample-metadata | Usable alone: No
SDRF template for non-human vertebrate samples (mammals, birds, fish, reptiles, amphibians). Must be combined with a technology template (ms-proteomics, affinity-proteomics, or ms-metabolomics).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
(override: requirement set to required) |
||
|
required |
Developmental stage of the organism |
ontology: efo |
adult, embryo, juvenile, larval stage |
|
recommended |
Strain or breed of the organism |
ontology: ncbitaxon |
C57BL/6, Sprague-Dawley, BALB/c, Wistar |
|
recommended |
Biological sex of the organism |
values: male, female, hermaphrodite |
14.7. invertebrates
Version: 1.1.0 | Layer: sample | Extends: sample-metadata | Usable alone: No
SDRF template for invertebrate samples (Drosophila, C. elegans, insects, etc.). Must be combined with a technology template (ms-proteomics, affinity-proteomics, or ms-metabolomics).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
(override: requirement set to required) |
||
|
required |
Developmental stage of the organism |
ontology: efo |
adult stage, larval stage, pupal stage, embryonic stage |
|
required |
Strain of the organism |
ontology: ncbitaxon |
Oregon-R, w1118, N2, Canton-S |
|
optional |
Genotype of the organism |
pattern: Genotype notation following standard conventions |
wild type, daf-2(e1370), w[*]; P{GAL4} |
14.8. plants
Version: 1.1.0 | Layer: sample | Extends: sample-metadata | Usable alone: No
SDRF template for plant samples (Arabidopsis, crops, etc.). Must be combined with a technology template (ms-proteomics, affinity-proteomics, or ms-metabolomics).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
ontology: uberon, bto, po |
flower bud, leaf, root, seed |
||
|
required |
(override: requirement set to required) |
||
|
required |
Developmental stage of the plant |
ontology: efo |
seedling stage, flowering stage, rosette growth stage, senescent stage |
|
recommended |
Cultivar, ecotype, or accession of the plant |
pattern: Plant cultivar or ecotype name |
Col-0, Ler-0, Nipponbare, B73 |
|
recommended |
Growth conditions for the plant |
pattern: Description of growth conditions |
long day (16h light/8h dark), short day (8h light/16h dark), continuous light, greenhouse |
|
recommended |
(override: requirement set to recommended) |
14.9. clinical-metadata
Version: 1.0.0 | Layer: sample | Extends: sample-metadata | Usable alone: No
SDRF template for clinical study samples with treatment, demographics, and lifestyle metadata. Applicable to any organism. Combine with organism template (human, vertebrates) and technology template (ms-proteomics, affinity-proteomics, or ms-metabolomics).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
(override: requirement set to required) |
||
|
optional |
Chemical compound or drug applied to sample |
ontology: chebi, ncit, efo |
doxorubicin, cisplatin, tamoxifen, metformin |
|
optional |
Dose or concentration of compound treatment |
number with unit (mg/kg, uM, nM, mg, ug, mg/mL, ug/mL, mM) |
10 mg/kg, 50 uM, 100 nM, 5 mg |
|
optional |
Duration of treatment exposure |
number with unit (hour, day, minute, week, month) |
24 hour, 5 day, 30 minute, 2 week |
|
optional |
Treatment status at time of sampling |
values: pre-treatment, on treatment, post-treatment, treatment naive |
|
|
optional |
Response to treatment (for studies measuring therapeutic outcomes) |
ontology: ncit |
complete response, partial response, progressive disease, stable disease |
|
optional |
Pre-existing medical conditions or comorbidities |
ontology: mondo, efo, doid |
diabetes mellitus, hypertension, obesity |
|
optional |
Body mass index (BMI) in kg/m^2 |
pattern: Numeric BMI value |
24.5, 31.2, 18.7 |
|
optional |
Patient smoking status |
ontology: ncit |
never smoker, former smoker, current smoker |
|
optional |
Menopausal status for female patients |
values: pre-menopausal, peri-menopausal, post-menopausal |
|
|
optional |
Method of genetic modification (knockout, knockdown, overexpression, transduction) |
ontology: efo |
knockout, knockdown, overexpression, transduction, … |
|
optional |
Observable characteristics or traits (drug sensitivity, molecular markers, expression phenotypes) |
ontology: pato, efo |
drug resistant, HER2-positive, high expresser, wild-type phenotype |
|
optional |
Body weight of the subject |
number with unit (kg, g, lb) |
70 kg, 55 kg, 154 lb |
|
optional |
Height of the subject |
number with unit (cm, m) |
175 cm, 1.75 m, 160 cm |
|
optional |
Specific anatomical location or context of sampling within the organism part |
ontology: uberon, bto |
tumor, normal tissue adjacent to tumor, left ventricle, frontal cortex |
|
optional |
Known genetic variant, mutation, or genotype of the subject |
pattern: Genotype as free text (gene name + variant) |
BRCA1 mutation carrier, KRAS G12D mutant, wild type, TP53 R175H |
14.10. oncology-metadata
Version: 1.0.0 | Layer: sample | Extends: clinical-metadata | Usable alone: No
SDRF template for cancer/oncology study samples with tumor staging, grading, and clinical outcome metadata. Extends clinical-metadata with oncology-specific columns. Combine with organism template (human, vertebrates) and technology template (ms-proteomics, affinity-proteomics, or ms-metabolomics).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
optional |
Disease progression stage (stage I-IV, chronic phase, end stage) |
ontology: ncit, efo |
stage I, stage II, stage III, stage IV, … |
|
optional |
Histological tumor grade (describes how abnormal cells look) |
ontology: ncit |
grade 1, grade 2, grade 3, grade 4, … |
|
optional |
TNM staging notation (describes extent of cancer spread) |
ontology: ncit |
T2N1M0, T3N0M0, T1N0M0, T4N2M1 |
|
optional |
Tumor size measurement |
number with unit (cm, mm) |
2.5 cm, 15 mm, 0.8 cm |
|
optional |
Tumor mass/weight measurement |
number with unit (g, mg) |
15 g, 250 mg |
|
optional |
Cancer molecular or histologic subtype |
ontology: ncit |
luminal A, luminal B, HER2-enriched, triple-negative, … |
|
optional |
Location where cancer has spread from primary site |
ontology: uberon, bto |
liver, lung, bone, brain |
|
optional |
Specific anatomical location of biopsy |
ontology: uberon, bto |
breast, colon, prostate, lung |
|
optional |
Free-text clinical details (receptor status, treatment history, surgical details) |
pattern: Free-text clinical data |
ER+/PR+/HER2-, prior chemotherapy with doxorubicin, surgical resection performed |
|
optional |
Relevant medical history information for the patient |
pattern: Free-text clinical history |
family history of breast cancer, previous radiation therapy, no significant medical history |
|
optional |
Patient survival time for survival analysis studies |
number with unit (month, year, day, week) |
24 month, 3 year, 180 day |
|
optional |
Time of last clinical follow-up for longitudinal studies |
number with unit (month, year, day, week) |
36 month, 5 year, 365 day |
|
optional |
Number of mitoses per high-power field (indicator of tumor proliferation) |
pattern: Mitotic rate as count or count per HPF |
5, 12/10 HPF, 3/10 HPF |
|
optional |
Dukes staging for colorectal cancer (A, B, C, D) |
values: A, B, C, D |
|
|
optional |
Ann Arbor staging for lymphoma (I, II, III, IV with optional A/B suffix) |
pattern: Ann Arbor stage (I-IV with optional A/B suffix for symptoms, E for extranodal, S for spleen) |
IA, IIB, IIIA, IVB, … |
|
optional |
Gleason score for prostate cancer grading (sum of two pattern grades, range 2-10) |
pattern: Gleason score as sum (e.g., 7) or component pattern (e.g., 3+4) |
7, 3+4, 4+3, 9, … |
|
optional |
Weiss scoring system for adrenal cortical carcinoma (low or high) |
values: low, high |
14.11. dia-acquisition
Version: 1.1.0 | Layer: experiment | Extends: ms-proteomics | Usable alone: No
SDRF template for Data-independent acquisition (DIA) experiments. Extends ms-proteomics with DIA-specific columns.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Mass spectrometry acquisition method (restricted to DIA for this template) |
single value only; values: Data-independent acquisition |
|
|
recommended |
Lower m/z limit of the DIA scan window |
pattern: m/z value as a number |
400, 350.5 |
|
recommended |
Upper m/z limit of the DIA scan window |
pattern: m/z value as a number |
1200, 1000 |
|
recommended |
Width of the isolation window in m/z units |
pattern: Width in m/z |
25, 8, 4 |
|
recommended |
Specific DIA method variant used |
ontology: pride |
SWATH-MS, MSE, All ion fragmentation, diaPASEF |
14.12. single-cell
Version: 1.0.0 | Layer: experiment | Extends: ms-proteomics | Usable alone: No
SDRF template for single-cell proteomics (SCP) experiments. Works with any organism - combine with appropriate sample template (human, vertebrates, invertebrates, or plants). Aligned with Nature Methods SCP guidelines (Gatto et al., 2023).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
recommended |
(override: requirement set to recommended) |
||
|
required |
Method used to isolate single cells (FACS, cellenONE, LCM, etc.) |
values: FACS, cellenONE, microfluidics, laser capture microdissection, … |
|
|
required |
Unique identifier for each single cell within the experiment. Required per SCP guidelines for tracking cells through analysis. |
identifier |
cell_001, SC_A1, well_B3, barcode_ATCGATCG, … |
|
recommended |
Batch identifier for sample preparation (plate, chip, processing batch). Critical for batch effect correction. |
||
|
recommended |
Number of cells per well/reaction. Use 1 for true single cells, higher numbers for small pools. |
pattern: Number of cells |
1, 5, 10, 100 |
|
recommended |
TMT/TMTpro channel used for the carrier proteome |
pattern: TMT channel label for carrier |
TMT131C, TMTpro134N, TMT126 |
|
recommended |
TMT/TMTpro channel used for the reference sample (for normalization across sets) |
pattern: TMT channel label for reference |
TMT131N, TMTpro133C, TMT127N |
|
optional |
Forward scatter (FSC) value from flow cytometry - proxy for cell size |
pattern: FSC value (numeric) |
316.0, 250 |
|
optional |
Side scatter (SSC) value from flow cytometry - proxy for cell granularity/complexity |
pattern: SSC value (numeric) |
301.0, 184 |
|
optional |
Markers used for cell sorting/enrichment with optional intensity values |
pattern: Enrichment marker(s) and optional intensity |
CD45+, GFP+, CD3+CD4+, CD34:APC-Cy7-A=276.0, … |
|
optional |
Viability status of the cell at isolation |
values: live, viable, dead, unknown |
|
|
optional |
Cell cycle phase if determined (e.g., by FACS or computational inference) |
values: G1, S, G2, G2/M, … |
|
|
optional |
Physical diameter of the isolated cell if measured (in micrometers) |
number with unit (um, μm) |
15 um, 20.5 um, 12 μm |
|
optional |
X,Y coordinates if cells were isolated from a spatial context (e.g., LCM from tissue) |
pattern: Spatial coordinates |
X=100;Y=250, X=50.5;Y=120.3 |
|
optional |
Tissue section identifier for spatially resolved single-cell proteomics |
pattern: Tissue section identifier |
section_001, slide_A_section_3 |
|
optional |
Nozzle diameter used for FACS-based single cell isolation (in micrometers) |
number with unit (um, μm) |
70 um, 100 um, 130 μm |
|
optional |
Sorting mode used during FACS isolation |
values: single cell, purity, yield, 4-way purity |
|
|
optional |
Type and manufacturer of the microfluidics chip used for single cell isolation |
pattern: Chip type/manufacturer identifier |
Fluidigm C1, Cellenion cellenCHIP, nanowell chip |
|
optional |
Model of the laser capture microdissection microscope used for cell isolation |
pattern: LCM microscope model name |
Leica LMD7, Zeiss PALM MicroBeam, Thermo LCM |
|
optional |
Version of the nanoPOTS chip used for single cell sample preparation |
pattern: nanoPOTS chip version identifier |
nanoPOTS v1, nanoPOTS v2, 9-well chip |
14.13. immunopeptidomics
Version: 1.0.0 | Layer: experiment | Extends: ms-proteomics | Usable alone: No
SDRF template for immunopeptidomics experiments (MHC-bound peptide identification). Works with any organism - combine with appropriate sample template (human for HLA typing, vertebrates for H-2/MHC typing in mouse, etc.).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
MHC protein complex targeted for immunopeptidome enrichment (GO:0042611) |
values: MHC class I protein complex, MHC class II protein complex, non-classical MHC protein complex, mutant MHC protein complex, MHC protein complex with serotype |
|
|
required |
Method used to enrich MHC-bound peptides |
values: immunoaffinity purification, immunoaffinity purification (iodoacetamide), mild acid elution, detergent lysis |
|
|
recommended |
MHC alleles expressed by the sample (PRIDE:0000893) following IPD-MHC nomenclature (https://www.ebi.ac.uk/ipd/mhc/). Use IPD-IMGT/HLA notation for human (HLA-A*02:01), H-2 notation for mouse (H-2Kb, H-2Db), or appropriate IPD-MHC notation for other species. Multiple alleles can be separated by semicolons. |
pattern: MHC allele notation (HLA for human, H-2 for mouse). Supports multi-allele (semicolon-separated), 2-4 field resolution. |
HLA-A*02:01, HLA-B*07:02, HLA-A*02:01;HLA-B*07:02;HLA-C*07:02, HLA-A*02:01:01, … |
|
optional |
MHC typing method used (PRIDE:0000894). Values mapped to NCIT where available: NGS-based typing (NCIT:C101293), sequence-based typing (NCIT:C130180), PCR-SSO (NCIT:C130181), PCR-SSP (NCIT:C130179), PCR-based genotyping (NCIT:C17003) |
values: NGS-based typing, sequence-based typing, PCR-SSO, PCR-SSP, … |
|
|
recommended |
Antibody clone used for MHC immunoprecipitation |
pattern: Antibody clone name |
W6/32, BB7.2 |
14.14. crosslinking
Version: 1.0.0 | Layer: experiment | Extends: ms-proteomics | Usable alone: No
SDRF template for crosslinking mass spectrometry (XL-MS) experiments. Extends ms-proteomics with crosslinking-specific columns for data analysis.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
recommended |
MS-based cross-linking methodology used to identify this as a crosslinking dataset |
values: cross-linking mass spectrometry |
|
|
recommended |
(override: requirement set to recommended) |
||
|
required |
Cross-linker compound with structured properties for analysis tools. Format: NT=name;AC=accession;CL=cleavable;TA=targets;MH/ML=stub masses Uses XLMOD ontology (parent term XLMOD:00004). |
structured_kv |
NT=DSS;AC=XLMOD:02001, NT=BS3;AC=XLMOD:02000, NT=DSSO;AC=XLMOD:02010;CL=yes;TA=K,S,T,Y,nterm;MH=54.01;ML=85.98, NT=EDC;AC=XLMOD:02009;CL=no;TA=K,D,E |
|
required |
Fragmentation method used in MS2. Critical for cleavable crosslinkers (DSSO, DSBU) which generate diagnostic stub ions under specific fragmentation conditions. |
ontology: ms, pride |
HCD, CID, ETD, EThcD, … |
|
recommended |
Collision energy used for fragmentation. Important for cleavable crosslinker analysis. |
pattern: Collision energy format: {value} {unit} where unit is NCE or eV. For stepped collision energies, use semicolon-separated values or 'stepped' prefix. |
30 NCE, 30% NCE, 27 eV, 25 NCE;27 NCE;30 NCE, … |
|
recommended |
Method used to enrich crosslinked peptides before MS analysis |
ontology: pride, ms |
size exclusion chromatography, strong cation exchange chromatography, high-pH reversed-phase chromatography, FAIMS |
|
optional |
Maximum Cα-Cα distance constraint provided by the crosslinker (for structural interpretation) |
number with unit (Å) |
30 Å, 26.4 Å, 11.4 Å |
|
optional |
Concentration of crosslinking reagent used |
number with unit (mM, uM, µM) |
2 mM, 500 uM, 1 mM |
|
optional |
Duration of the crosslinking reaction |
number with unit (min, h, s) |
30 min, 1 h, 45 min |
|
optional |
Temperature at which crosslinking was performed |
number with unit (°C) |
25°C, 4°C, 37°C, room temperature |
|
optional |
Molar ratio of crosslinker to protein |
pattern: Ratio format (e.g., 50:1 or 1:1 w/w) |
3001, 6001, 1:1 w/w |
|
optional |
Reagent used to quench the crosslinking reaction |
pattern: Chemical name of quenching reagent |
Tris-HCl, ammonium bicarbonate, glycine |
14.15. cell-lines
Version: 1.1.0 | Layer: experiment | Extends: sample-metadata | Usable alone: No
SDRF template for cell line samples with Cellosaurus-based annotation. Cell lines can originate from any organism - combine with appropriate organism template (human for HeLa, vertebrates for NIH 3T3, invertebrates for Sf9).
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Name of the cell line |
ontology: clo, bto, efo |
HeLa, HEK293, MCF7, A549 |
|
required |
Disease state of the donor tissue from which the cell line was established |
||
|
required |
Cellosaurus accession number for the cell line |
accession: cellosaurus |
CVCL_0030, CVCL_0004 |
|
recommended |
Official Cellosaurus name for the cell line |
||
|
optional |
Original source term, local provenance, or finer tissue-of-origin context for the cell line |
ontology: uberon, bto |
cervix, kidney, breast |
|
recommended |
Passage number of the cell line used in the experiment |
pattern: Passage number should be an integer or range |
10, 15-20, 5 |
|
optional |
BioBank or source from which the cell line was obtained |
pattern: Source of the cell line |
ATCC, DSMZ, ECACC, Sigma-Aldrich |
|
optional |
Method used to authenticate the cell line identity |
pattern: Authentication method used |
STR profiling, SNP fingerprinting, cytogenetic analysis |
|
recommended |
Culture medium used to grow the cell line |
ontology: ncit |
DMEM, RPMI 1640, MEM, Ham’s F-12 |
|
optional |
Developmental stage of the donor from which the cell line was derived |
ontology: efo |
adult, embryonic, fetal, neonatal |
|
optional |
Ancestry category of the cell line donor (if known) |
ontology: hancestro |
European, African, East Asian, South Asian |
|
recommended |
Storage temperature of the cell line (in Celsius) |
number with unit (°C) |
-80 °C, -20 °C, 4 °C |
14.16. metaproteomics
Version: 1.0.0 | Layer: sample | Extends: base | Usable alone: No
Base SDRF template for metaproteomics experiments (microbial community proteomics). Extends base directly and defines MIxS-aligned sample metadata. When combined with ms-proteomics, sample-metadata columns (organism, disease, cell type) are excluded. Use a child template (human-gut, soil, water) for environment-specific fields.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Type of environmental sample analyzed (ENVO or EFO term). Corresponds to MIxS env_medium (MIXS:0000014). |
ontology: envo, efo |
soil, seawater, gut microbiome, wastewater, … |
|
recommended |
Geographic location where sample was collected (GAZ term or coordinates). Corresponds to MIxS geo_loc_name (MIXS:0000010). |
ontology: gaz |
Pacific Ocean, Amazon rainforest, 47.6062 N, 122.3321 W |
|
recommended |
Environmental material from which the sample was obtained (ENVO term). Corresponds to MIxS env_medium (MIXS:0000014). |
ontology: envo |
soil, seawater, freshwater, feces, … |
|
optional |
Date when sample was collected (ISO 8601) |
date |
2024, 2024-01, 2024-01-15 |
|
optional |
Method used to collect the environmental sample |
pattern: Collection method description |
grab sample, core sample, swab, filtration |
|
optional |
Depth at which sample was collected. Corresponds to MIxS depth (MIXS:0000018). |
number with unit (m, cm, mm) |
10 m, 50 cm, 100 m |
|
optional |
Altitude or elevation of sampling site. Corresponds to MIxS elevation (MIXS:0000093). |
number with unit (m) |
500 m, 1200 m, 0 m |
|
optional |
Temperature at sampling location. Corresponds to MIxS temperature (MIXS:0000113). |
number with unit (°C) |
25 °C, 4 °C, -20 °C |
|
optional |
pH at sampling location |
pattern: pH value |
7.0, 5.5, 8.2 |
|
optional |
Storage conditions for the sample before analysis |
pattern: Storage conditions |
-80C, liquid nitrogen, 4C |
|
optional |
Accession number for matched metagenome data |
accession: |
MGYA00001234, SRP123456 |
|
optional |
Source of the microbiome being studied (e.g., gut microbiome, rhizosphere microbiome) |
pattern: Microbiome source description |
gut microbiome, rhizosphere microbiome, oral microbiome, skin microbiome |
|
optional |
Estimated microbial biomass in the sample |
pattern: Biomass estimation |
1e9 cells/g, high biomass, low biomass |
|
optional |
Level of host protein contamination if known |
pattern: Host contamination level |
low (<5%), moderate (5-20%), high (>20%) |
|
optional |
Contaminant database(s) used in database search |
pattern: Contaminant database name(s) |
cRAP, MaxQuant contaminants, cRAP;MaxQuant contaminants |
|
optional |
Identifier or name of mock community standard used |
pattern: Mock community identifier |
ZymoBIOMICS Microbial Community Standard, ATCC MSA-1000 |
|
optional |
Description of mock community composition (species and ratios) |
pattern: Community composition description |
8 bacteria + 2 yeasts at defined ratios, even mix of 10 species |
|
optional |
Semicolon-separated list of organisms expected in mock community |
pattern: Semicolon-separated organism list |
E. coli;B. subtilis;S. cerevisiae;L. fermentum, Bacillus subtilis;Staphylococcus aureus |
14.17. human-gut
Version: 1.0.0 | Layer: sample | Extends: metaproteomics | Usable alone: No
SDRF template for human gut metaproteomics. Extends metaproteomics with host-associated columns aligned with the GSC MIxS human-gut extension (0016004). Combine with ms-proteomics for MS acquisition columns.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Reference to the Sample. This term is used for cross-reference samples in SDRF for proteomics (MIXS:0001107). |
identifier |
Sample_1, Sample_2 |
|
required |
Host organism for host-associated microbiome samples |
ontology: ncbitaxon |
Homo sapiens |
|
recommended |
De-identified unique identifier for the host subject. Corresponds to MIxS host_subject_id (MIXS:0000861). |
identifier |
subject_001, patient_A, anonymized |
|
recommended |
Host disease diagnoses. Corresponds to MIxS host_disease_stat (MIXS:0000031). |
ontology: mondo, doid |
inflammatory bowel disease, colorectal cancer, healthy |
|
recommended |
Body site where sample was obtained. Corresponds to MIxS host_body_site (MIXS:0000867). |
ontology: uberon, bto |
stool, oral cavity, colon |
|
recommended |
Observed genotype MIXS:0000365. |
ontology: efo, gene |
homozygous, heterozygous |
|
recommended |
Phenotype of human or other host MIXS:0000274. |
ontology: pato, hp |
polydactyly, arachnodactyly |
|
optional |
Age of host at the time of sampling. Corresponds to MIxS host_age (MIXS:0000255). |
pattern: Age in standard format (Y=year, M=month, W=week, D=day, H=hour) |
45Y, 8W, 3M |
|
optional |
Sex of the host organism. Corresponds to MIxS host_sex (MIXS:0000811). |
values: male, female, intersex |
|
|
optional |
Body mass index (weight/height^2). Corresponds to MIxS host_body_mass_index (MIXS:0000317). |
pattern: BMI numeric value |
22.5, 30.1, 18.5 |
|
optional |
Height of the host. Corresponds to MIxS host_height (MIXS:0000264). |
number with unit (cm, m) |
175 cm, 1.75 m |
|
optional |
Total mass of the host. Corresponds to MIxS host_tot_mass (MIXS:0000263). |
number with unit (kg, g) |
70 kg, 85 kg |
|
optional |
Ethnicity of the host. Corresponds to MIxS ethnicity (MIXS:0000895). |
pattern: Ethnicity description |
European, East Asian, African |
|
optional |
Diet type of the host. Corresponds to MIxS host_diet (MIXS:0000869). |
pattern: Diet description |
omnivore, vegan, western diet, high-fiber |
|
optional |
Special dietary restrictions. Corresponds to MIxS special_diet (MIXS:0000905). |
pattern: Special diet description |
gluten-free, low FODMAP, ketogenic |
|
optional |
Content of last meal and time since feeding. Corresponds to MIxS host_last_meal (MIXS:0000870). |
pattern: Last meal description |
breakfast 4 hours prior, fasting 12 hours |
|
optional |
Relationships to other hosts in the same study; can include multiple relationships MIXS:0000872. |
pattern: family relationship description |
father, daughter |
|
optional |
Most frequent job performed by subject MIXS:0000896. |
pattern: occupation description |
teacher, engineer |
|
optional |
History of GI tract disorders. Corresponds to MIxS gastroint_disord (MIXS:0000280). |
pattern: GI disorder description |
Crohn’s disease, ulcerative colitis, irritable bowel syndrome, none |
|
optional |
History of liver disorders. Corresponds to MIxS liver_disord (MIXS:0000282). |
pattern: Liver disorder description |
none, fatty liver disease, hepatitis |
|
optional |
Recent antibiotic exposure of the host |
pattern: Antibiotic treatment description |
none, amoxicillin 7 days prior, broad-spectrum |
|
optional |
Medication codes (IHMC). Corresponds to MIxS ihmc_medication_code (MIXS:0000884). |
pattern: Medication code(s) |
none, A02BC01, N02BE01 |
|
optional |
Substance produced by the body where sample was obtained. Corresponds to MIxS host_body_product (MIXS:0000888). |
pattern: Body product description |
stool, mucus, saliva |
|
optional |
Core body temperature at sample collection. Corresponds to MIxS host_body_temp (MIXS:0000874). |
number with unit (°C) |
36.6 °C, 37.2 °C |
|
optional |
Type of perturbation applied. Corresponds to MIxS perturbation (MIXS:0000754). |
pattern: Perturbation description |
antibiotic administration, dietary intervention, none |
|
optional |
Chemical compounds administered to the host. Corresponds to MIxS chem_administration (MIXS:0000751). |
pattern: Chemical administration description |
metformin 500mg daily, probiotics |
|
optional |
Resting pulse, measured as beats per minutes (MIXS:0000333). |
number with unit (bpm) |
65 bpm, 72 bpm, 85 bpm |
|
optional |
The taxonomic name of the organism(s) found living in mutualistic, commensalistic, or parasitic symbiosis with the specific host (MIXS:0001298). |
ontology: |
|
|
optional |
Whether full medical history was collected (MIXS:0000897). |
pattern: Medical history performed description |
45 year old male with a 2 week history of stomach pain. |
|
optional |
Name of the project within which the sequencing was organized (MIXS:0000092). |
identifier |
Human_microbiome_project |
|
optional |
Temperature at which sample was stored (MIXS:0000110). |
number with unit (°C) |
-80 °C, -20 °C |
|
optional |
Location at which sample was stored (MIXS:0000755). |
pattern: storage location description |
freezer, room |
|
optional |
Duration for which the sample was stored (MIXS:0000116). |
pattern: storage duration description |
40 days, 6 months |
|
optional |
Volume (ml) or mass (g) of total collected sample processed for DNA extraction (MIXS:0000111). |
number with unit (ml,g) |
1 ml, 2 g |
|
optional |
Total cell count of any organism per gram, volume, or area of sample, including count (MIXS:0000103). |
number with unit (CFU/ml) |
500 CFU/ml, 2000 CFU/ml |
|
optional |
Oxygenation status of sample (MIXS:0000753). |
number with unit (mmHg) |
80 mmHg, 100 mmHg |
|
optional |
Any other measurement performed or parameter collected, that is not listed here (MIXS:0000752). |
pattern: miscellaneous parameter description |
none |
14.18. soil
Version: 1.0.0 | Layer: sample | Extends: metaproteomics | Usable alone: No
SDRF template for soil metaproteomics. Extends metaproteomics with soil-specific columns aligned with the GSC MIxS soil extension (0016012). Combine with ms-proteomics for MS acquisition columns.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Reference to the Sample. This term is used for cross-reference samples in SDRF for proteomics (MIXS:0001107). |
identifier |
Sample_1, Sample_2 |
|
required |
Name of the project within which the sequencing was organized (MIXS:0000092). |
identifier |
Bermuda_Atlantic_Time-series_Study, Metaproteomes_of_Seawater_Microbial_Communities |
|
recommended |
Soil classification type (ENVO term) |
ontology: envo |
sandy loam, clay, peat, silt |
|
optional |
Height of the sampling site above mean sea level. |
values: 10 feet, 30 feet, 2 cm |
|
|
optional |
The angle between ground surface and a horizontal line (MIxS:0000646). |
values: 10 %, 30 % |
|
|
optional |
The direction a slope faces (MIxS:0000647). |
values: 300 degrees, north west, south |
|
|
optional |
Cross-sectional position in the hillslope where sample was collected (MIxS:0001084). |
values: summit, shoulder, backslope |
|
|
optional |
Drainage classification from a standard system such as the USDA system (MIxS:0001085). |
values: well drained, poorly drained, very poorly drained |
|
|
optional |
Present state of sample site (MIXS:0001080). |
pattern: Land use type |
agricultural, forest, urban, grassland, … |
|
optional |
Current vegetation type at sampling site. Corresponds to MIxS cur_vegetation (MIXS:0000312). |
ontology: envo |
grassland, deciduous forest, cropland |
|
optional |
method used in vegetation classification (MIXS:0000314). |
values: vegetation survey, quadrant sampling |
|
|
optional |
Previous land use and dates (MIXS:0000315). |
values: agricultural land 15 years, grassland forest 5 years |
|
|
optional |
Reference or method used in determining previous land use (MIXS:0000316). |
values: land registry records, historical archives |
|
|
optional |
Crop rotation history. Corresponds to MIxS crop_rotation (MIXS:0000318). |
pattern: Crop rotation description |
corn-soybean rotation, wheat-fallow, continuous corn |
|
optional |
Addition of fertilizers, pesticides, etc. - amount and time of applications (MIXS:0000639). |
values: urea 2mg/L, 2020-01-01, superphosphate 5mg/L, 2019-02-19 |
|
|
optional |
tilling methods (MIXS:0001081). |
values: chisel, digging spade |
|
|
optional |
evidence of fire (MIXS:0001086). |
date |
09-10-2025, 10-02-2002 |
|
optional |
evidence of flooding (MIXS:0000319). |
date |
09-10-2025, 10-02-2002 |
|
optional |
events that may have affected microbial populations (MIXS:0000320). |
date |
09-10-2025, 10-02-2002 |
|
optional |
Soil horizon from which sample was collected |
values: O horizon, A horizon, B horizon, C horizon, … |
|
|
optional |
method used in determining the horizon (MIXS:0000321). |
pattern: Horizon method description |
USDA Field Book, FAO Guidelines for Soil Profile Description, none |
|
optional |
Soil classification from the FAO World soil distribution (MIxS:0001083) |
values: acrisols, calcisols, durisols, gleysols |
|
|
optional |
Link to digitized soil maps or other soil classification information (MIXS:0000329). |
pattern: URL, DOI, or PMID linking to classification information |
https://example.com/soil-map, doi:10.1000/xyz123, PMID:12345678 |
|
optional |
Soil classification based on local soil classification system (MIXS:0000330). |
pattern: Local soil classification term or description |
Chernozem, Podzol, Local classification type A |
|
optional |
Method used for local soil classification, including reference to the classification system or methodology applied (MIXS:0000331). |
pattern: Description or reference to the local soil classification method |
FAO soil classification guidelines, USDA Soil Taxonomy methodology, National soil classification system (UK) |
|
optional |
The relative proportion of different grain sizes of mineral particles in a soil, expressed as percentages of sand, silt, and clay, optionally including a textural class (MIXS:0000335). |
pattern: Soil texture expressed as percentages and/or textural class |
40% sand 40% silt 20% clay, 30-50 silty clay loam, silty clay loam |
|
optional |
Method used to determine soil texture, including reference to the analytical or classification approach applied.(MIXS:0000336) |
pattern: Description or reference to the soil texture determination method |
Hydrometer method, Laser diffraction particle size analysis, Pipette method |
|
optional |
Link to climate resource associated with the sampling site. (MIXS:0000328) |
pattern: URL, DOI, or PMID linking to climate information |
https://climate-data.org/location123, doi:10.1000/climate456, PMID:98765432 |
|
optional |
Mean annual temperature at the sampling site, typically expressed in degrees Celsius.(MIXS:0000135) |
values: |
12.5 oC, 25 oC |
|
optional |
Mean seasonal temperature range or average during a defined season at the sampling site. (MIXS:0000136) |
pattern: Seasonal temperature description or value |
summer 18°C, winter -2 to 5°C |
|
optional |
Total annual precipitation at the sampling site, typically measured in millimeters per year.(MIXS:0000140) |
numeric |
800/year, 1200.5/year |
|
optional |
Precipitation amount during a defined season at the sampling site. (MIXS:0000141) |
pattern: Seasonal precipitation description or value |
summer 120 mm, winter 300 mm |
|
optional |
Method used to determine pH in the sample.(MIXS:0001106) |
pattern: Description of pH measurement method |
pH measured in water (1:2.5 soil:solution ratio), pH measured in CaCl2 solution |
|
optional |
Organic matter content of the sample.(MIXS:0000204) |
number with unit () |
5.2 %, 12 % |
|
optional |
Total organic carbon content. Corresponds to MIxS tot_org_carb (MIXS:0000533). |
pattern: Total organic carbon with unit |
15.2 g/kg, 2.5 % |
|
optional |
Method used to determine total organic carbon.(MIXS:0000337) |
pattern: Description of TOC measurement method |
dry combustion method, Walkley-Black method |
|
optional |
Organic nitrogen content of the sample.(MIXS:0000205) |
number with unit () |
0.2 %, 1.5 % |
|
optional |
Total nitrogen content of the sample.(MIXS:0000530) |
number with unit () |
0.5 %, 2.1 % |
|
optional |
Method used to determine total nitrogen content.(MIXS:0000338) |
pattern: Description of nitrogen measurement method |
Kjeldahl method, Dumas combustion method |
|
optional |
Water content of soil sample. Corresponds to MIxS water_content (MIXS:0000185). |
pattern: Water content with unit |
25 %, 0.25 g/g |
|
optional |
Method used to determine soil water content.(MIXS:0000323) |
pattern: Description of water content measurement method |
gravimetric method, time-domain reflectometry |
|
optional |
Microbial biomass present in the sample.(MIXS:0000650) |
number with unit () |
250 mg/kg, 1200 mg/kg |
|
optional |
Method used to determine microbial biomass.(MIXS:0000339) |
pattern: Description of microbial biomass measurement method |
chloroform fumigation extraction, substrate-induced respiration |
|
optional |
Concentration of heavy metals in the sample, including elements such as lead, cadmium, mercury, and others.(MIXS:0000652) |
pattern: Heavy metal concentrations with units or description |
Pb 50 mg/kg; Cd 2 mg/kg, Hg 0.5 mg/kg |
|
optional |
Method used to determine heavy metal concentrations in the sample.(MIXS:0000343) |
pattern: Description of heavy metal measurement method |
ICP-MS analysis, Atomic absorption spectroscopy |
|
optional |
Aluminum saturation of the soil, typically expressed as a percentage of cation exchange capacity occupied by aluminum.(MIXS:0000607) |
number with unit () |
15 %, 45.5 % |
|
optional |
Method used to determine aluminum saturation in the sample.(MIXS:0000324) |
pattern: Description of aluminum saturation measurement method |
KCl extraction followed by titration, ICP-OES after extraction |
|
optional |
Size of mesh used to sieve the sample prior to analysis.(MIXS:0000322) |
pattern: Sieve size or description |
2 mm mesh, 500 µm sieve |
|
optional |
Volume or weight of sample used for DNA extraction.(MIXS:0000111) |
pattern: Sample amount with unit |
0.5 g, 10 mL |
|
optional |
Whether multiple DNA extracts were pooled prior to sequencing.(MIXS:0000325) |
pattern: Indication of DNA extract pooling |
yes, pooled from 3 replicates, no |
|
optional |
Storage conditions of the sample prior to analysis, including temperature and duration if known.(MIXS:0000327) |
pattern: Storage condition description |
-80°C for 6 months, 4°C short-term storage, room temperature |
|
optional |
Link to additional analysis results or external resources related to the sample.(MIXS:0000340) |
pattern: URL, DOI, or PMID linking to additional analysis |
https://example.com/analysis-results, doi:10.1000/analysis123, PMID:12345678 |
|
optional |
Any additional parameter not captured by other fields; should include a name and value.(MIXS:0000752) |
pattern: Parameter name and value in key:value format |
{'enzyme_activity': 'high'}, {'soil_color': 'dark brown'} |
14.19. water
Version: 1.0.0 | Layer: sample | Extends: metaproteomics | Usable alone: No
SDRF template for aquatic metaproteomics. Extends metaproteomics with water-specific columns aligned with the GSC MIxS water extension (0016014). Combine with ms-proteomics for MS acquisition columns.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Reference to the Sample. This term is used for cross-reference samples in SDRF for proteomics (MIXS:0001107). |
identifier |
Sample_1, Sample_2 |
|
required |
Name of the project within which the sequencing was organized (MIXS:0000092). |
identifier |
Bermuda_Atlantic_Time-series_Study, Metaproteomes_of_Seawater_Microbial_Communities |
|
recommended |
Ecological depth zone of the sampling site |
values: epipelagic, mesopelagic, bathypelagic, abyssopelagic, … |
|
|
recommended |
Height of the sampling site above mean sea level. |
number with unit (feet, cm) |
10 feet, 30 feet, 2 cm |
|
recommended |
Stage of tide (MIXS:0000750). |
values: high tide, low tide |
|
|
recommended |
Measurement of total depth of water column (MIXS:0000634). |
number with unit (m) |
35 m, 10 m, 200 m |
|
recommended |
Measured speed and direction of water flow |
number with unit (m/s) |
0.35 m/s, 0.10 m/s |
|
recommended |
Measurement of mean friction velocity (MIXS:0000498). |
number with unit (m/s) |
0.035 m/s, 0.010 m/s |
|
recommended |
Measurement of mean peak friction velocity (MIXS:0000502). |
number with unit (m/s) |
0.015 m/s, 0.045 m/s |
|
recommended |
Pressure to which the sample is subject to, in atmospheres (MIXS:0000412) |
number with unit (dbar) |
15 dbar, 25 dbar |
|
optional |
Salinity measurement. Corresponds to MIxS salinity (MIXS:0000183). |
pattern: Salinity value with unit or descriptive term |
35 PSU, freshwater, brackish |
|
optional |
Electrical conductivity of water sample. Corresponds to MIxS conduc (MIXS:0000544). |
pattern: Conductivity with unit |
450 uS/cm, 1.2 mS/cm |
|
optional |
Density of the sample, which is its mass per unit volume (aka volumetric mass density) (MIXS:0000435). |
number with unit (kg/m3) |
1022 kg/m3, 1050 kg/m3 |
|
optional |
Redox potential, measured relative to a hydrogen cell, indicating oxidation or reduction potential (MIXS:0000182). |
number with unit (V) |
1.2 V, 0.80 V |
|
optional |
Raw or converted fluorescence of water (MIXS:0000704). |
number with unit (RFU) |
1.2 RFU, 3.8 RFU |
|
optional |
Light intensity at sampling depth. Corresponds to MIxS light_intensity (MIXS:0000706). |
pattern: Light intensity with unit |
500 lux, 100 umol/m2/s |
|
optional |
Turbidity measurement. Corresponds to MIxS turbidity (MIXS:0000191). |
pattern: Turbidity with unit |
5.2 NTU, 12 FNU |
|
optional |
Visible waveband radiance and irradiance measurements in the water column (MIXS:0000703). |
number with unit (µmol) |
120 µmol, 15 µmol |
|
optional |
Measurement of photon flux (MIXS:0000725). |
number with unit (µmol) |
120 µmol, 15 µmol |
|
optional |
Nitrate concentration. Corresponds to MIxS nitrate (MIXS:0000425). |
number with unit (mg/L, umol/L) |
0.5 mg/L, 10 umol/L |
|
optional |
Nitrite concentration. Corresponds to MIxS nitrite (MIXS:0000426). |
number with unit (mg/L) |
0.5 mg/L, 1 mg/L |
|
optional |
Nitrogen concentration. Corresponds to MIxS nitrogen (MIXS:0000504). |
number with unit (mg/L) |
10 mg/L, 15 mg/L |
|
optional |
Ammonium concentration. Corresponds to MIxS ammonium (MIXS:0000427). |
number with unit (mg/L) |
1 mg/L, 1.5 mg/L |
|
optional |
Phosphate concentration. Corresponds to MIxS phosphate (MIXS:0000505). |
number with unit (mg/L, umol/L) |
0.1 mg/L, 1.5 umol/L |
|
optional |
Dissolved inorganic nitrogen concentration (MIXS:0000698). |
number with unit (mmol/L) |
761 mmol/L, 500 mmol/L |
|
optional |
total inorganic nitrogen concentration (MIXS:0000745). |
number with unit (mg/L) |
76 mg/L, 40 mg/L |
|
optional |
total dissolved nitrogen concentration (MIXS:0000744). |
number with unit (mg/L) |
76 mg/L, 40 mg/L |
|
optional |
Dissolved inorganic phosphorus concentration (MIXS:0000106). |
number with unit (mmol/L) |
76 mmol/L, 56 mmol/L |
|
optional |
total nitrogen concentration (MIXS:0000102). |
number with unit (mmol/L) |
76 mmol/L, 56 mmol/L |
|
optional |
total phosphorus concentration. Corresponds to MIxS total phosphorus (MIXS:0000117). |
number with unit (mg/L) |
0.03 mg/L, 0.05mg/L |
|
optional |
soluble reactive phosphorus concentration (MIXS:0000738). |
number with unit (mg/L) |
0.1 mg/L, 0.2 mg/L |
|
optional |
ssilicate concentration. Corresponds to MIxS(MIXS:0000184). |
number with unit (mmol/L) |
0.05 mmol/L, 0.08 mmol/L |
|
optional |
Alkalinity measurement. Corresponds to MIxS alkalinity (MIXS:0000421). |
number with unit (mg/L, meq/L) |
120 mg/L, 2.5 meq/L |
|
optional |
Method used for alkalinity measurement (MIXS:0000298). |
identifier |
titration |
|
optional |
Calcium concentration. Corresponds to MIxS(MIXS:0000421). |
number with unit (mmol/L) |
0.2 mmol/L, 2.5 mmol/L |
|
optional |
Magnesium concentration. Corresponds to MIxS(MIXS:0000431). |
number with unit (mmol/kg) |
25 mmol/kg, 52.8 mmol/kg |
|
optional |
Potassium concentration. Corresponds to MIxS(MIXS:0000430). |
number with unit (mg/L) |
463 mg/L, 300 mg/L |
|
optional |
Sodium concentration. Corresponds to MIxS(MIXS:0000428). |
number with unit (mg/L) |
10.5 mg/L, 12 mg/L |
|
optional |
Sodium concentration. Corresponds to MIxS(MIXS:0000429). |
number with unit (mg/L) |
5000 mg/L, 3500 mg/L |
|
optional |
Sulfate concentration. Corresponds to MIxS(MIXS:0000423). |
number with unit (mmol/L) |
5 mmol/L, 3.5 mmol/L |
|
optional |
Sulfide concentration. Corresponds to MIxS(MIXS:0000424). |
number with unit (mmol/L) |
2 mmol/L, 1.5 mmol/L |
|
optional |
Bromide concentration. Corresponds to MIxS(MIXS:0000176). |
number with unit (ppm) |
0.05 ppm, 0.01 ppm |
|
optional |
Measurement of atmospheric data; can include multiple data (MIXS:0001097). |
pattern: Diet description |
wind speed; 9 knots, humidity; 75% |
|
optional |
Dissolved organic carbon concentration. Corresponds to MIxS (MIXS:0000433) |
number with unit (mmol/L) |
197 mmol/L, 75 mmol/L |
|
optional |
Dissolved inorganic carbon concentration. Corresponds to MIxS (MIXS:0000434) |
number with unit (mmol/kg) |
2059 mmol/kg, 1075 mmol/kg |
|
optional |
Dissolved carbon dioxide concentration. Corresponds to MIxS (MIXS:0000436) |
number with unit (mg/L) |
5 mg/L, 10 mg/L |
|
optional |
Dissolved oxygen concentration. Corresponds to MIxS diss_oxygen (MIXS:0000119). |
pattern: Dissolved oxygen with unit or descriptive term |
8.5 mg/L, hypoxic, anoxic |
|
optional |
Dissolved hydrogen concentration. Corresponds to MIxS (MIXS:0000205). |
number with unit (mmol/L) |
0.3 mmol/L, 0.5 mmol/L |
|
optional |
Dissolved organic nitrogen concentration. Corresponds to MIxS (MIXS:0000162). |
number with unit (mmol/L) |
0.04 mmol/L, 0.05 mmol/L |
|
optional |
Organic carbon concentration. Corresponds to MIxS (MIXS:0000508). |
number with unit (ug/L) |
1.5 ug/L, 1 ug/L |
|
optional |
Organic matter concentration. Corresponds to MIxS (MIXS:0000204). |
number with unit (ug/L) |
1.75 ug/L, 1 ug/L |
|
optional |
Organic nitrogen concentration. Corresponds to MIxS (MIXS:0000205). |
number with unit (umol/L) |
4 umol/L, 1 umol/L |
|
optional |
Particulate organic carbon concentration. Corresponds to MIxS (MIXS:0000515). |
number with unit (umol/L) |
1.92 umol/L, 1 umol/L |
|
optional |
Particulate organic nitrogen concentration. Corresponds to MIxS (MIXS:0000719). |
number with unit (umol/L) |
0.3 umol/L, 0.5 umol/L |
|
optional |
Total particulate carbon content. Corresponds to MIxS (MIXS:0000747). |
number with unit (umol/L, ug/L) |
35 umol/L, 5 ug/L |
|
optional |
Carbon to nitrogen concentration. Corresponds to MIxS (MIXS:0000310). |
pattern: carbon nitrogen measurement value |
0.475, 0.34576 |
|
optional |
Suspended particulate matter concentration. Corresponds to MIxS (MIXS:0000741). |
number with unit (mg/L) |
0.5 mg/L, 1 mg/L |
|
optional |
Chlorophyll concentration if measured |
number with unit (ug/L, mg/L) |
2.5 ug/L, 0.1 mg/L |
|
optional |
Primary production measurement |
number with unit (mg/m3/day, g/m2/day) |
100 mg/m3/day, 10 g/m2/day |
|
optional |
bacterial production measurement (MIXS:0000683) |
number with unit (mg/m3/day) |
5 mg/m3/day, 10 mg/m3/day |
|
optional |
bacterial respiration measurement (MIXS:0000684) |
number with unit (ug/m3/day, umolO2/L/hr) |
300 umolO2/L/hr, 100 umolO2/L/hr |
|
optional |
bacterial carbon production measurement (MIXS:0000173) |
number with unit (ug/L/hr) |
2.53 ug/L/hr, 1.23 ug/L/hr |
|
optional |
Aminopeptidase activity measurement (MIXS:0000172) |
number with unit (mol/L/hr) |
0.253 mol/L/hr, 0.123 mol/L/hr |
|
optional |
glucosidase activity measurement (MIXS:0000137) |
number with unit (mol/L/hr) |
5 mol/L/hr, 3 mol/L/hr |
|
optional |
phaeopigments concentration (MIXS:0000180) |
number with unit (mg/m3) |
2.5 mg/m3, 3 mg/m3 |
|
optional |
Total cell count of any organism (MIXS:0000174) |
number with unit (ton, kg, g) |
25 g, 3 kg |
|
optional |
phaeopigments concentration (MIXS:0000103) |
number with unit (CFU/ml) |
500 CFU/ml, 2000 CFU/ml |
|
optional |
Oxygenation status of sample (MIXS:0000753). |
number with unit (mmHg) |
80 mmHg, 100 mmHg |
|
optional |
Alkyl diethers concentration (MIXS:0000490). |
number with unit (mol/L) |
0.005 mol/L, 0.003 mol/L |
|
optional |
Diether lipids concentration (MIXS:0000178). |
number with unit (ng/L) |
0.5 ng/L, 0.3 ng/L |
|
optional |
Bishomohopanol concentration (MIXS:0000175). |
number with unit (ug/L, ug/g) |
14 ug/L, 4 ug/g |
|
optional |
Phospholipid fatty acid concentration (MIXS:0000181). |
pattern: Phospholipid fatty acid name and measurement value |
Linoleic acid; 2.8 mg/L, Palmitic acid; 1.5 mg/L |
|
optional |
Petroleum hydrocarbon concentration (MIXS:0000516). |
number with unit (umol/L) |
0.05 umol/L, 0.02 umol/L |
|
optional |
Temperature at which sample was stored (MIXS:0000110). |
number with unit (°C) |
-80 °C, -20 °C |
|
optional |
Location at which sample was stored (MIXS:0000755). |
pattern: storage location description |
freezer, room |
|
optional |
Duration for which the sample was stored (MIXS:0000116). |
pattern: storage duration description |
40 days, 6 months |
|
optional |
Volume or weight of sample used for DNA extraction.(MIXS:0000111) |
pattern: Sample amount with unit |
0.5 g, 10 mL |
|
optional |
mesh/pore size used to pre-filter/pre-sort the sample (MIXS:0000735). |
number with unit (um) |
0.1 um, 0.2 um |
|
optional |
Mesh or pore size of the device used to retain the sample (MIXS:0000736). |
number with unit (um) |
10 um, 20 um |
|
optional |
Type of perturbation applied. Corresponds to MIxS perturbation (MIXS:0000754). |
pattern: Perturbation description |
antibiotic administration, dietary intervention, none |
|
optional |
Any additional parameter not captured by other fields; should include a name and value.(MIXS:0000752) |
pattern: Parameter name and value in key:value format |
{'enzyme_activity': 'high'}, {'soil_color': 'dark brown'} |
14.20. gc-ms-metabolomics
Version: 1.0.0-dev | Layer: experiment | Extends: ms-metabolomics | Usable alone: No
SDRF template for GC-MS-based metabolomics experiments. Extends ms-metabolomics with derivatization and GC-specific columns.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
(override: requirement set to required) |
single value only; values: GC-MS-based metabolomics |
|
|
required |
(override: requirement set to required) |
values: electron ionization, chemical ionization, electron impact ionization |
|
|
required |
Derivatization protocol applied to the sample prior to GC injection. Use |
pattern: Free-text derivatization protocol |
MSTFA, MOX + MSTFA, BSTFA, methoximation followed by trimethylsilylation, … |
|
recommended |
Chemical reagent(s) used for derivatization |
ontology: chebi |
N-methyl-N-(trimethylsilyl)trifluoroacetamide, N,O-bis(trimethylsilyl)trifluoroacetamide, methoxyamine hydrochloride |
|
recommended |
GC column used for separation, including stationary phase and dimensions |
pattern: Free-text GC column description |
DB-5MS 30 m x 0.25 mm x 0.25 um, HP-5MS 30 m x 0.25 mm x 0.25 um, Rxi-5Sil MS 30 m x 0.25 mm x 0.25 um |
|
optional |
Carrier gas used in the GC system |
values: helium, hydrogen, nitrogen |
|
|
optional |
Free-text description of the GC oven temperature program |
pattern: Free-text oven program description |
60C 1 min, ramp 10C/min to 300C, hold 5 min, 70C 2 min, ramp 5C/min to 320C, hold 10 min |
14.21. lc-ms-metabolomics
Version: 1.0.0-dev | Layer: experiment | Extends: ms-metabolomics | Usable alone: No
SDRF template for LC-MS-based metabolomics experiments. Extends ms-metabolomics with chromatography-specific columns.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
(override: requirement set to required) |
single value only; values: LC-MS-based metabolomics |
|
|
required |
Chromatography mode used for separation. Should be a CHMO term (e.g. CHMO:0002302 reversed-phase chromatography, CHMO:0001025 hydrophilic interaction chromatography). |
pattern: Chromatography type as a CHMO-style ontology term (NT=…;AC=CHMO:NNNNNNN) or one of the common short names. Strict CHMO ontology validation is a follow-up. |
NT=reversed-phase chromatography;AC=CHMO:0002302, NT=hydrophilic interaction chromatography;AC=CHMO:0001025, reversed-phase chromatography, hydrophilic interaction chromatography |
|
recommended |
LC column used for separation, including manufacturer, model, and dimensions |
pattern: Free-text chromatography column description |
Waters ACQUITY HSS T3 100 x 2.1 mm 1.8 um, Phenomenex Kinetex C18 150 x 2.1 mm 2.6 um, SeQuant ZIC-pHILIC 150 x 2.1 mm 5 um |
|
optional |
Composition of mobile phase A |
pattern: Free-text composition of mobile phase A |
0.1% formic acid in water, 10 mM ammonium acetate in water pH 9.0, 5 mM ammonium formate in water |
|
optional |
Composition of mobile phase B |
pattern: Free-text composition of mobile phase B |
0.1% formic acid in acetonitrile, 10 mM ammonium acetate in 90% acetonitrile pH 9.0, methanol |
|
optional |
Free-text description of the chromatographic gradient program |
pattern: Free-text gradient description |
5% B 0-1 min, ramp to 95% B at 15 min, hold 3 min, re-equilibrate, isocratic 50% B for 10 min, 0-100% B in 30 min |
|
optional |
Mobile-phase flow rate |
number with unit (nL/min, uL/min, mL/min) |
300 nL/min, 250 uL/min, 0.4 mL/min |
14.22. ms-metabolomics
Version: 1.0.0-dev | Layer: technology | Extends: sample-metadata | Usable alone: Yes
Base SDRF template for mass spectrometry-based metabolomics. This is the parent template for LC-MS and GC-MS metabolomics experiments.
| Column Name | Req. | Description | Validators | Examples |
|---|---|---|---|---|
|
required |
Type of technology used |
single value only; values: metabolite profiling by mass spectrometry, LC-MS-based metabolomics, GC-MS-based metabolomics |
|
|
required |
Mass spectrometer instrument used |
ontology: psi-ms, pride |
Q Exactive, Xevo G2-S QTof, Orbitrap Fusion Lumos, timsTOF Pro |
|
required |
Ion source used for ionization (e.g. ESI, APCI, EI, MALDI). Should be a child of MS:1000008. |
ontology: psi-ms |
electrospray ionization, atmospheric pressure chemical ionization, electron ionization, matrix-assisted laser desorption ionization |
|
required |
Scan polarity (positive scan, negative scan, or polarity switching). Polarity switching is reserved for true in-method switching producing a single file; otherwise use two rows per sample (one per polarity). |
single value only; values: positive scan, negative scan, polarity switching |
|
|
required |
Mass spectrometry acquisition method (DDA, DIA, SIM, MRM, full scan) |
ontology: pride |
data-dependent acquisition, data-independent acquisition, selected reaction monitoring, parallel reaction monitoring, … |
|
recommended |
Pointer to the metabolite assignment file (MAF) for this assay. Per-assay constant — the same value is repeated across all rows of the same assay. Mirrors the MetaboBank semantics of Comment[Metabolite Assignment File]. |
pattern: Path or filename of the MAF file |
m_MTBLS1129_LC-MS_positive_reverse-phase_metabolite_profiling_v2_maf.tsv, maf_pos_lipidomics.tsv |
|
recommended |
Sample extraction method used to obtain the metabolite fraction |
pattern: Free-text extraction method, optionally CHMO-style NT=…;AC=CHMO:NNNNNNN |
methanol-water extraction, MTBE extraction, Bligh-Dyer extraction, Folch extraction, … |
|
optional |
Solvent or solvent mixture used for metabolite extraction |
ontology: chebi |
methanol, chloroform, methyl tert-butyl ether, acetonitrile |
|
recommended |
Class of metabolites targeted by the extraction and acquisition method |
values: polar metabolites, lipids, amino acids, fatty acids, … |
|
|
optional |
Internal standard(s) spiked into the sample for normalization or retention-time calibration |
ontology: chebi |
caffeine-d9, L-tryptophan-d5, palmitic acid-d31 |
|
recommended |
Type of biological matrix used as input (e.g. serum, plasma, CSF, urine, tissue extract) |
ontology: uberon, bto |
serum, plasma, cerebrospinal fluid, urine, … |
|
optional |
Batch identifier for sample preparation (plate, chip, processing batch). Useful for batch-effect correction. |
pattern: Sample preparation batch identifier |
plate1, batch_20220601, prep_A |
|
optional |
Liquid chromatography batch identifier for batch-effect tracking (e.g. column changes, LC system swaps) |
pattern: LC batch identifier |
LC1, column_A |
|
optional |
Date of MS data acquisition (ISO 8601 format recommended). Useful for tracking instrument drift and batch effects. |
pattern: Acquisition date/time |
2022-06-01, 2022-06-01T18:28:37 |
|
optional |
Mass analyzer used for MS2 acquisition |
ontology: psi-ms |
orbitrap, ion trap, TOF |
|
optional |
MS method-defined minimum precursor (MS1) m/z setting used to acquire the data |
m/z value |
100m/z, 200m/z |
|
optional |
MS method-defined maximum precursor (MS1) m/z setting used to acquire the data |
m/z value |
1200m/z, 1600m/z |
|
optional |
m/z scan range for MS1 spectra as an interval. Alternative to separate ms min mz / ms max mz columns. |
m/z range interval |
70m/z-1000m/z, 100m/z-1500m/z |
|
optional |
m/z scan range for MS2 spectra as an interval. Alternative to separate ms2 min mz / ms2 max mz columns. |
m/z range interval |
50m/z-1000m/z, 100m/z-1500m/z |
|
optional |
Precursor mass tolerance for feature detection / database search |
number with unit (ppm, Da, mmu) |
5 ppm, 10 ppm, 0.01 Da |
|
optional |
Fragment mass tolerance for feature detection / database search |
number with unit (ppm, Da, mmu) |
0.02 Da, 20 ppm |
|
optional |
Collision energy used for fragmentation |
pattern: Collision energy format: {value} {unit} where unit is NCE or eV. For multiple values, use semicolon-separated entries. |
30 NCE, 25 eV, 25 NCE;27 NCE;30 NCE |
|
optional |
MD5 integrity hash for the raw data file referenced in comment[data file]. Mirrors MetaboBank Comment[Raw Data File md5]. |
pattern: 32-character hex MD5 hash |
d41d8cd98f00b204e9800998ecf8427e, 9e107d9d372bb6826bd81d3542a419d6 |
|
optional |
Pointer to the processed data file (e.g. peak table, feature matrix). Mirrors MetaboBank Comment[Processed Data File]. |
pattern: Path or filename of the processed data file |
features_pos.tsv, peak_table_lipidomics.csv |
|
optional |
MD5 integrity hash for the processed data file. Mirrors MetaboBank Comment[Processed Data File md5]. |
pattern: 32-character hex MD5 hash |
d41d8cd98f00b204e9800998ecf8427e |
|
optional |
MD5 integrity hash for the metabolite assignment file. Mirrors MetaboBank Comment[Metabolite Assignment File md5]. |
pattern: 32-character hex MD5 hash |
d41d8cd98f00b204e9800998ecf8427e |
15. Intellectual Property Statement
The PSI takes no position regarding the validity or scope of any intellectual property or other rights that might be claimed to pertain to the implementation or use of the technology described in this document or the extent to which any license under such rights might or might not be available; neither does it represent that it has made any effort to identify any such rights. Copies of claims of rights made available for publication and any assurances of licenses to be made available or the result of an attempt made to obtain a general license or permission for the use of such proprietary rights by implementers or users of this specification can be obtained from the PSI Chair.
The PSI invites any interested party to bring to its attention any copyrights, patents or patent applications, or other proprietary rights which may cover technology that may be required to practice this recommendation. Please address the information to the PSI Chair (see contacts information at PSI website).
16. Copyright Notice
Copyright © Proteomics Standards Initiative (2020). All Rights Reserved.
This document and translations of it may be copied and furnished to others, and derivative works that comment on or otherwise explain it or assist in its implementation may be prepared, copied, published, and distributed, in whole or in part, without the restriction of any kind, provided that the above copyright notice and this paragraph are included on all such copies and derivative works. However, this document itself may not be modified in any way, such as by removing the copyright notice or references to the PSI or other organizations, except as needed for the purpose of developing Proteomics Recommendations in which case the procedures for copyrights defined in the PSI Document process must be followed, or as required to translate it into languages other than English.
The limited permissions granted above are perpetual and will not be revoked by the PSI or its successors or assigns.
This document and the information contained herein is provided on an "AS IS" basis and THE PROTEOMICS STANDARDS INITIATIVE DISCLAIMS ALL WARRANTIES, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO ANY WARRANTY THAT THE USE OF THE INFORMATION HEREIN WILL NOT INFRINGE ANY RIGHTS OR ANY IMPLIED WARRANTIES OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE."
17. How to cite
Please cite this document as:
Dai C, Füllgrabe A, Pfeuffer J, Solovyeva EM, Deng J, Moreno P, Kamatchinathan S, Kundu DJ, George N, Fexova S, Grüning B, Föll MC, Griss J, Vaudel M, Audain E, Locard-Paulet M, Turewicz M, Eisenacher M, Uszkoreit J, Van Den Bossche T, Schwämmle V, Webel H, Schulze S, Bouyssié D, Jayaram S, Duggineni VK, Samaras P, Wilhelm M, Choi M, Wang M, Kohlbacher O, Brazma A, Papatheodorou I, Bandeira N, Deutsch EW, Vizcaíno JA, Bai M, Sachsenberg T, Levitsky LI, Perez-Riverol Y. A proteomics sample metadata representation for multiomics integration and big data analysis. Nat Commun. 2021 Oct 6;12(1):5854. doi: 10.1038/s41467-021-26111-3. PMID: 34615866; PMCID: PMC8494749. [Manuscript - https://www.nature.com/articles/s41467-021-26111-3]
References
-
[1] Y. Perez-Riverol, S. European Bioinformatics Community for Mass, Toward a Sample Metadata Standard in Public Proteomics Repositories, J Proteome Res 19(10) (2020) 3906-3909. doi:10.1021/acs.jproteome.0c00376
-
[2] A. Gonzalez-Beltran, E. Maguire, S.A. Sansone, P. Rocca-Serra, linkedISA: semantic representation of ISA-Tab experimental metadata, BMC Bioinformatics 15 Suppl 14 (2014) S4. doi:10.1186/1471-2105-15-S14-S4
-
[3] T.F. Rayner, P. Rocca-Serra, P.T. Spellman, et al., A simple spreadsheet-based, MIAME-supportive format for microarray data: MAGE-TAB, BMC Bioinformatics 7 (2006) 489. doi:10.1186/1471-2105-7-489
-
[4] P. Blainey, M. Krzywinski, N. Altman, Points of significance: replication, Nat Methods 11(9) (2014) 879-80. doi:10.1038/nmeth.3091
-
[5] D. Gupta, I. Liyanage, Y. Perez-Riverol, et al., BioSamples database: the global hub for sample metadata and multi-omics integration, Nucleic Acids Res (2025). doi:10.1093/nar/gkaf1133