1. Status of this document
This document provides information to the proteomics community about a proposed standard for sample metadata annotations in public repositories called Sample and Data Relationship Format (SDRF)-Proteomics. Distribution is unlimited.
Version v1.1.0 - 2026-01
2. Abstract
The Human Proteome Organisation (HUPO) Proteomics Standards Initiative (PSI) defines community standards for data representation in proteomics to facilitate data comparison, exchange, and verification. This document presents a specification for the Sample and Data Relationship Format (SDRF-Proteomics).
Further detailed information, including any updates to this document, implementations, and examples is available at SDRF GitHub Repository. The official PSI web page for the document is: HUPO-PSI SDRF.
3. Motivation
Public proteomics data is valuable, but sample metadata is often missing or stored inconsistently across repositories (e.g., CPTAC uses Excel files, ProteomicsDB captures minimal properties) [1]. This heterogeneity prevents reproducibility and cross-dataset integration.
SDRF-Proteomics addresses this by providing a standard tab-delimited format to capture (Figure 1):
-
Sample metadata and characteristics
-
Data file acquisition parameters
-
Sample-to-file relationships (experimental design)

Figure 1: SDRF-Proteomics captures sample information and its relationship to data files.
The format is fully compatible with MAGE-TAB SDRF, enabling integration with transcriptomics metadata standards.
4. Specification structure
SDRF-Proteomics uses a two-tier system: this core specification defines the format rules, and templates provide metadata checklists for specific experiment types (Figure 2). Templates are organized in the templates/ directory, each with documentation and example files.
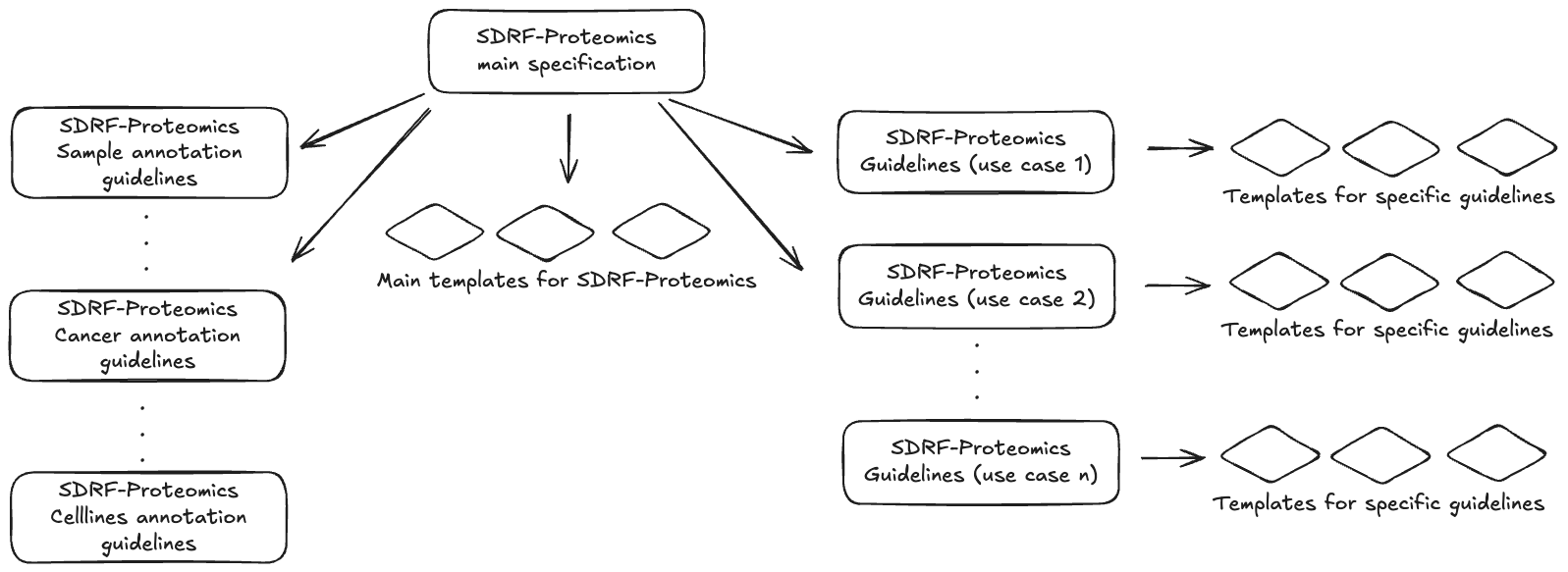
Figure 2: SDRF-Proteomics specification structure. The main specification defines the core rules and is extended by sample templates (human, vertebrates, etc.) and experiment-type templates (crosslinking, immunopeptidomics, etc.).
The official repository is GitHub, where you can find annotated example projects and the official validator sdrf-pipelines.
|
Important
|
Throughout this specification, the keywords "MUST", "REQUIRED", "SHOULD", "RECOMMENDED", and "OPTIONAL" are interpreted as described in RFC 2119. |
5. The SDRF-Proteomics Format
SDRF-Proteomics is a tab-delimited file where:
-
Each row = one sample linked to one data file
-
Each column = a property (sample characteristic, data file attribute, or factor value)
-
Each cell = the property value for that sample/file or a factor value.
Here’s a minimal example:
| source name | characteristics[organism] | characteristics[organism part] | characteristics[disease] | characteristics[biological replicate] | assay name | technology type | comment[proteomics data acquisition method] | comment[label] | comment[instrument] | comment[cleavage agent details] | comment[fraction identifier] | comment[technical replicate] | comment[data file] | factor value[disease] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
sample_1 |
homo sapiens |
liver |
normal |
1 |
run_1 |
proteomic profiling by mass spectrometry |
Data-dependent acquisition |
label free sample |
Q Exactive HF |
NT=Trypsin;AC=MS:1001251 |
1 |
1 |
sample_1.raw |
normal |
sample_2 |
homo sapiens |
liver |
hepatocellular carcinoma |
1 |
run_2 |
proteomic profiling by mass spectrometry |
Data-dependent acquisition |
label free sample |
Q Exactive HF |
NT=Trypsin;AC=MS:1001251 |
1 |
1 |
sample_2.raw |
hepatocellular carcinoma |
sample_3 |
homo sapiens |
not available |
not available |
1 |
run_3 |
proteomic profiling by mass spectrometry |
Data-dependent acquisition |
label free sample |
Q Exactive HF |
NT=Trypsin;AC=MS:1001251 |
1 |
1 |
sample_3.raw |
not available |
The file is organized into three column sections:
-
Sample metadata (
characteristics[…]) - organism, disease, tissue, etc. -
Data file metadata (
comment[…]) - instrument, label, fraction, data file -
Factor values (
factor value[…]) - variables under study for statistical analysis
|
Note
|
|
5.1. Versioning
The SDRF-Proteomics specification uses Semantic Versioning (MAJOR.MINOR.PATCH). Version numbers are prefixed with "v" (e.g., v1.1.0). Changes are proposed via GitHub pull requests to the dev branch.
For the complete versioning strategy — including template versioning, ontology updates, the deprecation policy, transition timelines, and migration tooling — see Versioning and Deprecation Policy.
5.2. Format rules
-
Case sensitivity: Text values are case-insensitive, but column names are case-sensitive. Use lowercase for all column names (e.g.,
source name,characteristics[organism],comment[label]). Incorrect casing likeSource NameorCharacteristics[organism]will cause validation failures. -
Space sensitivity: The SDRF is sensitive to spaces in column names (
sourcename≠source name). Column names must include appropriate spaces (e.g.,source name, notsourcename) but must NOT have a space before the bracket (e.g.,characteristics[organism], notcharacteristics [organism]). -
Column order: The SDRF columns follows some structure; first the sample metadata columns in Chapter 7; then the data file metadata columns in Chapter 8; followed by the factor values columns in [study-variables].
-
Extension: The extension of the SDRF file SHOULD be sdrf.tsv (preferred) or .txt.
5.3. Reserved words
There are general scenarios where cell values cannot be provided with actual data. The following reserved words MUST be used in these cases:
-
not available: In some cases, the column is mandatory in the format, but for some samples the corresponding value is unknown or could not be determined. In those cases, users SHOULD use not available.
-
not applicable: In some cases, the column is mandatory, but for some samples the corresponding value or concept does not apply. In those cases, users SHOULD use not applicable.
-
anonymized: In some cases, the value exists but has been intentionally redacted for privacy protection (e.g., in clinical studies with de-identified patient data). In those cases, users SHOULD use anonymized.
-
pooled: In some cases, the sample is a pool of multiple samples (e.g., TMT reference channels), and the value cannot be represented as a single value. In those cases, users SHOULD use pooled.
| Term | Meaning | Example | Use Case |
|---|---|---|---|
not available |
Value exists but is unknown or could not be determined |
characteristics[age] = not available |
Patient age was not recorded in the study |
not applicable |
Value or concept does not apply to this sample |
characteristics[age] = not applicable |
Synthetic peptide library has no age |
anonymized |
Value exists but is redacted for privacy protection |
characteristics[age] = anonymized |
Clinical study with de-identified patient data |
pooled |
Value represents a mixture of multiple samples |
characteristics[biological replicate] = pooled |
TMT reference channel pooled from multiple replicates |
5.4. SDRF file-level metadata
Since version 1.1.0, SDRF-Proteomics supports file-level metadata using dedicated columns. These columns provide information about the SDRF file itself, such as the specification version, template(s) used, annotation tool, and validation status. This column-based approach maintains compatibility with spreadsheet applications (Excel, Google Sheets) and existing data processing tools.
The following metadata columns are supported:
| Column | Description | Example Value | Requirement | Ontology Term |
|---|---|---|---|---|
|
SDRF-Proteomics specification version used. Should follow semantic versioning format (vMAJOR.MINOR.PATCH) |
v1.1.0 |
RECOMMENDED |
PRIDE:0000839 |
|
Template name and version used for annotation. Two formats are supported: simple format ( |
human v1.1.0 or NT=human;VV=v1.1.0 |
OPTIONAL |
PRIDE:0000832 |
|
Software tool, script, or method used to generate or annotate the SDRF file. Two formats are supported: simple format ( |
lesSDRF v0.1.0 or NT=lesSDRF;VV=v0.1.0 |
OPTIONAL |
PRIDE:0000840 |
|
Cryptographic hash (e.g., SHA-256) generated after successful validation |
sha256:abc123… |
OPTIONAL |
PRIDE:0000834 |
|
Note
|
When combining multiple templates (e.g., human + ms-proteomics), use multiple comment[sdrf template] columns, one per template. The value in each row should be identical for all samples in the file.
|
Example of an SDRF file with metadata columns (simplified example showing only select columns; see Chapter 10 for complete required columns):
| source name | characteristics[organism] | characteristics[disease] | assay name | comment[data file] | comment[sdrf version] | comment[sdrf template] | comment[sdrf template] | comment[sdrf annotation tool] |
|---|---|---|---|---|---|---|---|---|
| sample_1 | homo sapiens | normal | run_1 | sample_1.raw | v1.1.0 | human v1.1.0 | ms-proteomics v1.1.0 | lesSDRF v0.1.0 |
| sample_2 | homo sapiens | breast cancer | run_2 | sample_2.raw | v1.1.0 | human v1.1.0 | ms-proteomics v1.1.0 | lesSDRF v0.1.0 |
5.5. Table Column headers
Depending on each section the column headers (property names) will be prefixed with the following prefixes:
-
characteristics: Sample metadata (e.g. characteristics[organism]) -
comment: Data file metadata (e.g. comment[data file]) -
factor value: Factor values properties (e.g. factor value[disease])
Each property name MUST be a valid ontology term or a valid controlled vocabulary term. Each section will have some specific order for column headers.
|
Note
|
A list of all controlled vocabularies and ontologies supported are in the Chapter 12 section. On each section we also provide a list of properties that are supported. |
5.6. Table Cell values
The value for each property, (e.g. characteristics, comment, factor value) corresponding to each sample or data file can be represented in multiple ways.
-
Free Text (Human readable): In the free text representation, the value is provided as text without Ontology support (e.g. colon or providing accession numbers). This is only RECOMMENDED when the text inserted in the table is the exact name of an ontology/CV term in EFO. If the term is not in EFO, other ontologies can be used.
| source name | characteristics[organism] |
|---|---|
sample 1 |
homo sapiens |
sample 2 |
homo sapiens |
-
Ontology url (Computer readable): Users can provide the corresponding URI (Uniform Resource Identifier) of the ontology/CV term as a value. This is recommended for enriched files where the user does not want to use intermediate tools to map from free text to ontology/CV terms.
-
Key=value representation (Human and Computer readable): The current representation aims to provide a mechanism to represent the complete information of the ontology/CV term including Accession, Name and other additional properties. In the key=value pair representation, the Value of the property is represented as an Object with multiple properties, where the key is one of the properties of the object and the value is the corresponding value for the particular key. An example of key value pairs is post-translational modification (see Protein Modifications):
NT=Glu->pyro-Glu;MT=fixed;PP=Anywhere;AC=Unimod:27;TA=E
6. Validating SDRF Files
The official validator for SDRF-Proteomics files is sdrf-pipelines, a Python tool that checks your SDRF file for errors and compliance with the specification.
Installation:
pip install sdrf-pipelinesBasic Validation:
# Validate an SDRF file
parse_sdrf validate-sdrf --sdrf_file your_file.sdrf.tsv
# Validate with a specific template
parse_sdrf validate-sdrf --sdrf_file your_file.sdrf.tsv --template humanFor more information, visit: sdrf-pipelines on GitHub
7. SDRF-Proteomics: Samples metadata
The Sample metadata section provides information about the samples of origin and their characteristics. Each sample contains a source name (unique identifier) and a set of characteristics columns. The first column of the file should be the source name and the following columns should be the characteristics of the sample. For example, for any proteomics experiment (human, vertebrate, cell line), the following characteristics should be provided:
-
source name: Unique sample name (it can be present multiple times if the same sample is used several times in the same dataset)
-
characteristics[organism]: The organism of the Sample of origin. Values MUST come from NCBI Taxonomy.
-
characteristics[organism part]: The part of organism’s anatomy or substance arising from an organism from which the biomaterial was derived (e.g., liver). Values SHOULD come from UBERON or BTO.
-
characteristics[disease]: The disease under study in the Sample. Values SHOULD come from MONDO, EFO, or DOID. For healthy/control samples, use
normal(PATO:0000461) - see Disease Annotation Guidelines. -
characteristics[cell type]: A cell type is a distinct morphological or functional form of cell (e.g., epithelial, glial). Values SHOULD come from Cell Ontology (CL) or BTO.
Example:
| source name | characteristics[organism] | characteristics[organism part] | characteristics[disease] | characteristics[cell type] |
|---|---|---|---|---|
sample_treat |
homo sapiens |
liver |
liver cancer |
not available |
sample_control |
homo sapiens |
liver |
liver cancer |
not available |
|
Note
|
|
7.1. BioSamples database integration
Use the OPTIONAL characteristics[biosample accession number] column to link samples to BioSamples [5], enabling cross-database integration with genomics and transcriptomics data. Formats: SAMN* (NCBI) or SAMEA* (EBI).
7.2. Encoding sample technical and biological replicates
SDRF-Proteomics uses two REQUIRED columns to track replicates [4]:
-
characteristics[biological replicate]: Independent biological samples. Numbering restarts per experimental condition (factor value group).
-
comment[technical replicate]: Repeated measurements of the same sample (e.g., multiple injections)
When no replicates are performed, set both columns to 1. For pooled samples, use pooled for biological replicate.
| source name | characteristics[biological replicate] | comment[fraction identifier] | comment[technical replicate] | comment[data file] |
|---|---|---|---|---|
patient_001 |
1 |
1 |
1 |
P001_F1_TR1.raw |
patient_001 |
1 |
1 |
2 |
P001_F1_TR2.raw |
patient_002 |
2 |
1 |
1 |
P002_F1_TR1.raw |
patient_002 |
2 |
1 |
2 |
P002_F1_TR2.raw |
7.3. Pooled samples
When multiple samples are pooled into one (e.g., TMT/iTRAQ reference channels for normalization), use the characteristics[pooled sample] column to indicate pooling status. Allowed values:
-
not pooled: Regular individual samples
-
pooled: Sample is pooled but individual sources are unknown
-
SN=sample1;SN=sample2;…: Lists source names of pooled samples when known
Example:
| source name | characteristics[pooled sample] | characteristics[organism] | characteristics[age] | comment[label] | comment[data file] |
|---|---|---|---|---|---|
sample_1 |
not pooled |
homo sapiens |
45Y |
TMT126 |
file01.raw |
sample_2 |
not pooled |
homo sapiens |
52Y |
TMT127N |
file01.raw |
pooled_ref |
SN=sample_1;SN=sample_2 |
homo sapiens |
pooled |
TMT131C |
file01.raw |
|
Tip
|
For pooled samples, use pooled for individual-specific fields (biological replicate, age, sex) to indicate a mixture rather than a single sample.
|
7.4. Sample Metadata Guidelines
For detailed guidance on annotating sample metadata, refer to the following conventions documents:
-
Sample Metadata Guidelines - Detailed guidelines for age, sex, disease, organism part, cell type, developmental stage, spiked-in samples, and other sample characteristics
-
Human Sample Metadata Guidelines - Human-specific metadata including disease staging, treatment history, demographics, and lifestyle factors
8. SDRF-Proteomics: data files metadata
The connection between samples and data files is done using properties annotated with the comment prefix. All properties referring to a data file (e.g., MS run file) are annotated with the category comment. This differentiates data file properties from sample properties (characteristics).
8.1. CV Term Format for Data File Metadata
For data file metadata (comment columns) that reference ontology terms, use the structured format: NT={term name};AC={accession}
Examples: NT=HCD;AC=PRIDE:0000590, NT=Orbitrap;AC=MS:1000484
This format enables automated validation and software extraction from raw files. Sample metadata (characteristics) can use simple term names since they are typically human-annotated.
The following properties MUST be provided for each data file in mass spectrometry-based proteomics experiments. For affinity-based proteomics (Olink, SomaScan), see the Affinity-Proteomics template for different required columns.
| Column | Requirement | Description | Ontology |
|---|---|---|---|
|
REQUIRED |
Unique identifier for an MS run/data file |
Free text |
|
REQUIRED |
Technology used to capture the data |
Fixed values |
|
REQUIRED |
DDA, DIA, PRM, SRM |
PRIDE:0000659 |
|
REQUIRED |
Label applied to sample (or "label free sample") |
PRIDE - Labels |
|
REQUIRED |
Mass spectrometer model |
PSI-MS - Instruments |
|
REQUIRED |
Enzyme information (use "not applicable" for top-down/undigested samples) |
PSI-MS - Cleavage agents |
|
REQUIRED |
Fraction number (1 if not fractionated) |
Integer |
|
REQUIRED |
Technical replicate number (1 if none) |
Integer |
|
REQUIRED |
Name of the raw file |
Free text |
Example:
| source name | assay name | technology type | comment[proteomics data acquisition method] | comment[label] | comment[instrument] | comment[data file] |
|---|---|---|---|---|---|---|
sample_1 |
sample1_run1 |
proteomic profiling by mass spectrometry |
data-dependent acquisition |
label free sample |
Q Exactive HF |
sample1.raw |
8.2. Sample Preparation and Fragmentation (MS-based only)
|
Note
|
This section applies to mass spectrometry-based proteomics experiments only. For affinity-based proteomics, these properties do not apply. |
For detailed documentation of sample preparation and MS/MS fragmentation properties, see the MS-Proteomics Template:
-
Sample preparation: depletion, reduction reagent, alkylation reagent
-
Fractionation: fractionation method (used with
comment[fraction identifier]) -
Fragmentation: collision energy, dissociation method
8.3. Proteomics data acquisition method
Proteomics data acquisition method can happen in multiple ways: Data Dependent Acquisition (DDA), Data Independent Acquisition (DIA), and targeted approaches. The SDRF-Proteomics file format REQUIRES capturing the method used for the data acquisition in the comment[proteomics data acquisition method] column. The values MUST be children of the PRIDE ontology term proteomics data acquisition method (PRIDE:0000659). The following values are commonly used:
|
Important
|
The comment[proteomics data acquisition method] column is REQUIRED for all mass spectrometry-based SDRF files. This field must be explicitly specified and cannot be omitted or assumed. |
You can find an example of a DIA experiment in the following link: DIA example
|
Tip
|
For DIA experiments, additional properties like MS1 scan range can be captured. See DIA Scan Window Limits in the MS-Proteomics Template. |
8.4. MS-Proteomics Template
For detailed guidance on data file metadata, refer to the conventions document:
-
MS-Proteomics Template - Detailed guidelines for labels, instruments, modifications, cleavage agents, mass tolerances, RAW file URIs, and other data file properties
9. Additional SDRF Rules
9.1. Column Cardinality
Some columns can appear multiple times for the same sample. The cardinality rules are:
-
Single (1): Column appears exactly once per sample (e.g.,
characteristics[biological replicate]) -
Multiple (*): Column can appear multiple times (e.g.,
comment[modification parameters]can specify multiple post-translational modifications)
Example of multiple comment[modification parameters] columns:
| source name | characteristics[…] | comment[modification parameters] | comment[modification parameters] | … |
|---|---|---|---|---|
sample-1 |
… |
NT=Carbamidomethyl;AC=UNIMOD:4;TA=C;MT=fixed;PP=Anywhere |
NT=Oxidation;AC=UNIMOD:35;TA=M;MT=variable;PP=Anywhere |
… |
9.2. Row Uniqueness Requirements
Uniqueness constraints ensure data integrity:
-
MUST be unique (error):
source name+assay name+comment[label] -
SHOULD be unique (warning):
source name+assay name -
Assay name: Each data file MUST have a unique
assay name
|
Note
|
For multiplexed experiments (TMT, iTRAQ), multiple rows share the same assay name since samples are in one MS run. The comment[label] distinguishes samples within the run.
|
10. Templates
A template is a predefined set of metadata columns that ensures consistent annotation for specific experiment types. Templates define REQUIRED, RECOMMENDED, and OPTIONAL columns to make datasets FAIR-compliant.
10.1. Template Architecture
Templates follow a layered hierarchy:
| Layer | Templates | Description |
|---|---|---|
TECHNOLOGY (required) |
Minimum valid SDRF - choose one |
|
SAMPLE (recommended) |
Organism-specific metadata |
|
EXPERIMENT (optional) |
cell-lines, crosslinking, dda-acquisition, dia-acquisition, single-cell, immunopeptidomics |
Methodology-specific columns |
Child templates inherit all columns from parents and may add new columns or strengthen requirements (e.g., optional → required).
10.2. Specifying Templates in SDRF Files
Declare templates using comment[sdrf template] columns. Only list leaf templates (parents are implied). When using multiple templates, add multiple columns with the same name. Two formats are supported:
-
Simple format (preferred):
template_name vX.Y.Z -
Key=value format:
NT=template_name;VV=vX.Y.Z
source name ... comment[sdrf template] comment[sdrf template]
sample_1 ... human v1.1.0 crosslinking v1.0.0Common examples:
| Experiment Type | Template Columns |
|---|---|
Human MS proteomics |
|
Mouse MS proteomics |
|
Human crosslinking |
Two columns: |
Human Olink |
Two columns: |
10.3. Available Templates
Sample templates (organism-specific):
| Template | Use For | Key Columns |
|---|---|---|
Human clinical samples |
disease, age, sex, ancestry |
|
Mouse, rat, zebrafish |
disease, developmental stage, strain |
|
Drosophila, C. elegans |
disease, developmental stage, genotype |
|
Arabidopsis, crops |
disease, developmental stage, growth conditions |
Experiment-type templates:
-
DDA Acquisition - dissociation method, collision energy, modifications
-
DIA Acquisition - scan windows, isolation width, spectral library
-
Cell Lines - Cellosaurus integration
-
Single-Cell - cell isolation, carrier proteome
-
Immunopeptidomics - MHC class, HLA typing
-
Crosslinking MS - crosslinker reagents
-
Metaproteomics - environmental sample type
Download templates from the templates folder.
10.4. Extending Templates
You can add custom columns beyond template requirements for study-specific metadata. Rules:
-
Use
characteristics[…]for sample metadata,comment[…]for technical metadata -
Column names MUST be valid ontology terms (search OLS)
-
Use controlled vocabularies for values when available
See Common Additional Columns and SDRF Terms Reference for commonly used columns.
10.5. Contributing New Templates
To propose a new template, open an issue on GitHub and submit a pull request.
11. Factor Values (Study Variables)
Factor values identify the experimental variables being studied - the conditions you want to compare in your analysis. They highlight which sample characteristics are the focus of your experiment.
11.1. Column Format
factor value[{variable name}]
11.2. When to Use Factor Values
Use factor values to indicate:
-
The primary variable(s) under investigation
-
Conditions being compared (e.g., disease vs. normal, treated vs. untreated)
-
Variables that define experimental groups
|
Note
|
Use normal (not "control") in the disease field for healthy samples. "Control" is an experimental design concept, not a disease state. See Disease Annotation Guidelines for details.
|
11.3. Rules
-
Factor value columns SHOULD appear after all characteristics and comment columns
-
Multiple factor values can be used when studying multiple variables
-
The value in a factor value column typically mirrors a characteristics column value
11.4. Example
In an experiment comparing tumor vs. normal tissue across different cancer stages:
| source name | … | characteristics[disease] | characteristics[disease staging] | … | factor value[disease] | factor value[disease staging] |
|---|---|---|---|---|---|---|
tumor_sample_1 |
… |
breast carcinoma |
stage II |
… |
breast carcinoma |
stage II |
normal_sample_1 |
… |
normal |
not applicable |
… |
normal |
not applicable |
tumor_sample_2 |
… |
breast carcinoma |
stage III |
… |
breast carcinoma |
stage III |
In this example, both disease and disease staging are factor values because the experiment aims to compare expression differences between disease states and across cancer stages.
12. Ontologies and Controlled Vocabularies
SDRF-Proteomics uses ontologies and controlled vocabularies (CVs) to standardize metadata values. The following ontologies are supported:
| Category | Ontology/CV | Description | Notes |
|---|---|---|---|
General Purpose |
|||
General |
General experimental metadata |
||
General |
Phenotype and Trait Ontology |
||
General |
Biomedical terminology |
||
General |
Proteomics-specific terms |
||
Organism and Taxonomy |
|||
Taxonomy |
Organism classification |
||
Anatomy and Cell Types |
|||
Anatomy |
Cross-species anatomy ontology |
||
Cell Type |
Cell type classification |
||
Anatomy |
Tissues and cell lines |
||
Anatomy |
Plant anatomy and development |
For plant samples |
|
Anatomy |
Drosophila anatomy |
For Drosophila samples |
|
Anatomy |
C. elegans anatomy |
For C. elegans samples |
|
Anatomy |
Zebrafish anatomy and development |
For zebrafish samples |
|
Disease (see Disease Annotation Guidelines) |
|||
Disease |
Unified disease ontology |
RECOMMENDED |
|
Disease |
Disease terms from EFO |
||
Healthy samples |
Use |
||
Cell Lines |
|||
Cell Lines |
Cell line knowledge resource |
RECOMMENDED |
|
Cell Lines |
Cell line ontology |
||
Mass Spectrometry and Proteomics |
|||
MS/Proteomics |
Instruments, methods, parameters |
||
Modifications |
Protein modifications database |
||
Modifications |
Protein modifications ontology |
||
Other |
|||
Chemistry |
Chemical Entities of Biological Interest |
||
Environment |
Environmental sample classification |
For metaproteomics |
|
Ancestry |
Human ancestry categories |
For human samples |
|
13. Examples of Annotated Datasets
The following table provides links to example SDRF files for different experiment types. Click "View in Explorer" to open the SDRF file in the interactive viewer.
| Experiment Type | Dataset | Description | View | Source |
|---|---|---|---|---|
Label-free |
PXD008934 |
Human proteome label-free quantification |
||
TMT |
PXD017710 |
TMT-labeled quantitative proteomics |
||
SILAC |
PXD000612 |
SILAC-based quantification |
||
DIA |
PXD018830 |
Data-independent acquisition |
||
Phosphoproteomics |
PXD000759 |
PTM enrichment study |
||
Cell lines |
PXD001819 |
Cell line proteomics |
|
Tip
|
Use the SDRF Explorer to browse all {total_datasets}+ annotated datasets with filtering, statistics, and interactive viewing. |
A comprehensive collection of annotated projects is available at: Annotated Projects Repository
14. Intellectual Property Statement
The PSI takes no position regarding the validity or scope of any intellectual property or other rights that might be claimed to pertain to the implementation or use of the technology described in this document or the extent to which any license under such rights might or might not be available; neither does it represent that it has made any effort to identify any such rights. Copies of claims of rights made available for publication and any assurances of licenses to be made available or the result of an attempt made to obtain a general license or permission for the use of such proprietary rights by implementers or users of this specification can be obtained from the PSI Chair.
The PSI invites any interested party to bring to its attention any copyrights, patents or patent applications, or other proprietary rights which may cover technology that may be required to practice this recommendation. Please address the information to the PSI Chair (see contacts information at PSI website).
15. Copyright Notice
Copyright © Proteomics Standards Initiative (2020). All Rights Reserved.
This document and translations of it may be copied and furnished to others, and derivative works that comment on or otherwise explain it or assist in its implementation may be prepared, copied, published, and distributed, in whole or in part, without the restriction of any kind, provided that the above copyright notice and this paragraph are included on all such copies and derivative works. However, this document itself may not be modified in any way, such as by removing the copyright notice or references to the PSI or other organizations, except as needed for the purpose of developing Proteomics Recommendations in which case the procedures for copyrights defined in the PSI Document process must be followed, or as required to translate it into languages other than English.
The limited permissions granted above are perpetual and will not be revoked by the PSI or its successors or assigns.
This document and the information contained herein is provided on an "AS IS" basis and THE PROTEOMICS STANDARDS INITIATIVE DISCLAIMS ALL WARRANTIES, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO ANY WARRANTY THAT THE USE OF THE INFORMATION HEREIN WILL NOT INFRINGE ANY RIGHTS OR ANY IMPLIED WARRANTIES OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE."
16. How to cite
Please cite this document as:
Dai C, Füllgrabe A, Pfeuffer J, Solovyeva EM, Deng J, Moreno P, Kamatchinathan S, Kundu DJ, George N, Fexova S, Grüning B, Föll MC, Griss J, Vaudel M, Audain E, Locard-Paulet M, Turewicz M, Eisenacher M, Uszkoreit J, Van Den Bossche T, Schwämmle V, Webel H, Schulze S, Bouyssié D, Jayaram S, Duggineni VK, Samaras P, Wilhelm M, Choi M, Wang M, Kohlbacher O, Brazma A, Papatheodorou I, Bandeira N, Deutsch EW, Vizcaíno JA, Bai M, Sachsenberg T, Levitsky LI, Perez-Riverol Y. A proteomics sample metadata representation for multiomics integration and big data analysis. Nat Commun. 2021 Oct 6;12(1):5854. doi: 10.1038/s41467-021-26111-3. PMID: 34615866; PMCID: PMC8494749. [Manuscript - https://www.nature.com/articles/s41467-021-26111-3]
References
-
[1] Y. Perez-Riverol, S. European Bioinformatics Community for Mass, Toward a Sample Metadata Standard in Public Proteomics Repositories, J Proteome Res 19(10) (2020) 3906-3909. doi:10.1021/acs.jproteome.0c00376
-
[2] A. Gonzalez-Beltran, E. Maguire, S.A. Sansone, P. Rocca-Serra, linkedISA: semantic representation of ISA-Tab experimental metadata, BMC Bioinformatics 15 Suppl 14 (2014) S4. doi:10.1186/1471-2105-15-S14-S4
-
[3] T.F. Rayner, P. Rocca-Serra, P.T. Spellman, et al., A simple spreadsheet-based, MIAME-supportive format for microarray data: MAGE-TAB, BMC Bioinformatics 7 (2006) 489. doi:10.1186/1471-2105-7-489
-
[4] P. Blainey, M. Krzywinski, N. Altman, Points of significance: replication, Nat Methods 11(9) (2014) 879-80. doi:10.1038/nmeth.3091
-
[5] D. Gupta, I. Liyanage, Y. Perez-Riverol, et al., BioSamples database: the global hub for sample metadata and multi-omics integration, Nucleic Acids Res (2025). doi:10.1093/nar/gkaf1133